Chemiosmotic coupling hypothesis
Definition noun A theory postulated by the biochemist Peter Mitchell in 1961 to describe ATP synthesis by way of a proton electrochemical coupling . Accordingly, hydrogen ion s ( proton s) are pumped from the mitochondrial matrix to the intermembrane space via the hydrogen carrier protein s while the electrons are transferred along the electron transport chain in the mitochondrial inner membrane. As the hydrogen ions accumulate in the intermembrane space, an energy-rich proton gradient is established. As the proton gradient becomes sufficiently intense the hydrogen ion s tend to diffuse back to the matrix (where hydrogen ion s are less) via the ATP synthase (a transport protein). As the hydrogen ion s diffuse (through the ATP synthase ) energy is released which is then used to drive the conversion of ADP to ATP (by phosphorylation ). Supplement This theory was not previously well accepted until a great deal of evidence for proton pumping by the complexes of the electron transfer chain emerged. This began to favor the chemiosmotic hypothesis, and in 1978, Peter Mitchell was awarded the Nobel Prize in Chemistry. Also called: chemiosmotic hypothesis See also: chemiosmosis , mitochondrion
Last updated on February 24th, 2022

You will also like...

Passive and Active Types of Immunity
Lymphocytes are a type of white blood cell capable of producing a specific immune response to unique antigens. In thi..

Vascular Plants: Ferns and Relatives
Ferns and their relatives are vascular plants, meaning they have xylem and phloem tissues. Because of the presence of va..

Freshwater Communities & Lentic Waters
Lentic or still water communities can vary greatly in appearance -- from a small temporary puddle to a large lake. The s..

Sigmund Freud and Carl Gustav Jung
In this tutorial, the works of Carl Gustav Jung and Sigmund Freud are described. Both of them actively pursued the way h..

Biological Cell Defense
Organisms employ different strategies to boost its defenses against antigens. Humans have an immune system to combat pat..

Population Regulation in an Ecosystem
With regard to the population size of a species and what factors may affect them, two factors have been defined. They ar..
Related Articles...

No related articles found
If you're seeing this message, it means we're having trouble loading external resources on our website.
If you're behind a web filter, please make sure that the domains *.kastatic.org and *.kasandbox.org are unblocked.
To log in and use all the features of Khan Academy, please enable JavaScript in your browser.
AP®︎/College Biology
Course: ap®︎/college biology > unit 3.
- First Law of Thermodynamics introduction
- Second Law of Thermodynamics and entropy
- The laws of thermodynamics
- Reaction coupling to create glucose-6-phosphate
ATP and reaction coupling
- Introduction to metabolism: Anabolism and catabolism
- Overview of metabolism
- Cellular energy
Introduction
Atp structure and hydrolysis, hydrolysis of atp, reaction coupling, atp in reaction coupling, case study: let's make sucrose.
- In the first reaction, a phosphate group is transferred from ATP to glucose, forming a phosphorylated glucose intermediate (glucose-P). This is an energetically favorable (energy-releasing) reaction because ATP is so unstable, i.e., really "wants" to lose its phosphate group.
- In the second reaction, the glucose-P intermediate reacts with fructose to form sucrose. Because glucose-P is relatively unstable (thanks to its attached phosphate group), this reaction also releases energy and is spontaneous.
Different types of reaction coupling in the cell
Case study: sodium-potassium pump.
- Three sodium ions bind to the sodium-potassium pump, which is open to the interior of the cell.
- The pump hydrolyzes ATP, phosphorylating itself (attaching a phosphate group to itself) and releasing ATP. This phosphorylation event causes a shape change in the pump, in which it closes off on the inside of the cell and opens up to the exterior of the cell. The three sodium ions are released, and two potassium ions bind to the interior of the pump.
- The binding of the potassium ions triggers another shape change in the pump, which loses its phosphate group and returns to its inward-facing shape. The potassium ions are released into the interior of the cell, and the pump cycle can begin again.
Attribution:
Works cited:.
- Reece, J. B., Urry, L. A., Cain, M. L., Wasserman, S. A., Minorsky, P. V., and Jackson, R. B. (2011). The regeneration of ATP. In Campbell biology (10th ed., pp. 151). San Francisco, CA: Pearson.
- Berg, J. M., Tymoczsko, J. L., and Stryer, L. (2002). A thermodynamically unfavorable reaction can be driven by a favorable reaction. In Biochemistry (5th ed, section 14.1.1). New York, NY: W.H. Freeman. Retrieved from http://www.ncbi.nlm.nih.gov/books/NBK22439/#_A1943_ .
- Singh, N. K. (2007). ATP is the main energy currency in cells. In BIOL 1020 lecture notes: Chapter 8 . Retrieved from http://www.auburn.edu/academic/classes/biol/1020/singh/lecturenotes/ .
- Solomon, E., Martin, C., Martin, D., and Berg, L. (2014). ATP donates energy through the transfer of a phosphoric group. In Biology (10th ed., p. 154). Boston, MA: Cengage Learning.
Additional references:
Want to join the conversation.
- Upvote Button navigates to signup page
- Downvote Button navigates to signup page
- Flag Button navigates to signup page


An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- v.9(4); 2019 Apr
An update of the chemiosmotic theory as suggested by possible proton currents inside the coupling membrane
Alessandro maria morelli.
1 Pharmacy Department, Biochemistry Lab, University of Genova, Viale Benedetto XV 3, 16132 Genova, Italy
Silvia Ravera
2 Experimental Medicine Department, University of Genova, Via De Toni 14, 16132 Genova, Italy
Daniela Calzia
Isabella panfoli, associated data.
This article has no additional data.
Understanding how biological systems convert and store energy is a primary purpose of basic research. However, despite Mitchell's chemiosmotic theory, we are far from the complete description of basic processes such as oxidative phosphorylation (OXPHOS) and photosynthesis. After more than half a century, the chemiosmotic theory may need updating, thanks to the latest structural data on respiratory chain complexes. In particular, up-to date technologies, such as those using fluorescence indicators following proton displacements, have shown that proton translocation is lateral rather than transversal with respect to the coupling membrane. Furthermore, the definition of the physical species involved in the transfer (proton, hydroxonium ion or proton currents) is still an unresolved issue, even though the latest acquisitions support the idea that protonic currents, difficult to measure, are involved. Moreover, F o F 1 -ATP synthase ubiquitous motor enzyme has the peculiarity (unlike most enzymes) of affecting the thermodynamic equilibrium of ATP synthesis. It seems that the concept of diffusion of the proton charge expressed more than two centuries ago by Theodor von Grotthuss is to be taken into consideration to resolve these issues. All these uncertainties remind us that also in biology it is necessary to consider the Heisenberg indeterminacy principle, which sets limits to analytical questions.
1. Introduction
The ‘chemiosmotic theory’ formulated by Mitchell [ 1 ], a researcher with an Anglo-Saxon training in chemistry, dates back more than 50 years. The theory has universally been accepted, although it immediately raised several controversies, which lasted until today. An upgrading of the chemiosmotic theory appears necessary in light of the enormous progress of bioanalytic techniques defining the fine structure of the macromolecular complexes involved in oxidative phosphorylation (OXPHOS) [ 2 – 7 ], notably studies on complex I (NADH: ubiquinone oxidoreductase) and complex IV (cytochrome c oxidase) [ 2 , 3 , 8 ]. These data allow further insight into the proton pathway, a key issue of the theory. The actual proton path across the membrane, the putative proton concentrations on either sides of the membrane and the consequent membrane potential have been the subjects of countless studies. In all evidence, it appears that a free proton osmosis would be impossible, it being a quantum particle that binds to water forming hydronium ions (H 3 O + ). In fact, any free proton in the membrane would quickly be drained by the aqueous phase, releasing the energy associated with the solvation process, to the detriment of the membrane. Also, free protons display a huge destructive force on any biological membrane they pass through. Accordingly, some membrane transporters (such as the potential-dependent proton pump Hv1) are designed specifically to prevent the proton destructive force [ 9 ]. Also, a number of reports argue that protons accumulating onto the respiring membrane never reside in the aqueous phase [ 10 – 13 ].
Therefore, in this work, we hypothesize that the coupling modality between the electron transport chain and the protonic movement could happen inside the membrane to prevent a proton release, opening new scenarios to explain the basic mechanisms of aerobic metabolism. In other words, in this review, we debate the possibility to update the chemiosmotic theory and unravel the role of local processes in the coupling. This may help in developing new strategies for innovative research centred on cellular bioenergetics.
2. The chemiosmotic theory and F o F 1 -ATP synthase
The basic formulation of Mitchell's theory is schematically depicted in figure 1 , where ATP synthase was also indicated, differently from the original release of 1961, where necessarily it was not depicted. At the time ATP synthesis was attributed to the membrane as a whole, in the form of a generic subtraction of H + and OH − to ADP and orthophosphate to form ATP.

Schematic of the 1961 Mitchell chemiosmotic theory. A delocalized coupling is depicted among protons extruded by the electron transport chain (ETC) and ATP synthesis. The overall process is arbitrarily divided in the two phases: the ‘RedOx coupling’, in which the proton movement is operated by the ETC, and the ‘proton coupling’, in which proton movement is coupled with ATP synthesis, by F o F 1 -ATP synthase.
The experimental data in support of the theory came successively and are reported in literature as a huge amount of contributions. Reviews have been published and we refer to them for a complete documentation [ 14 , 15 ], only the most significant issues being mentioned here. Particularly influential were the data produced in 1966 by A. T. Jagendorf & E. Uribe in the famous ‘acid bath experiment’ [ 16 ]. They obtained an ATP synthesis inducing a transmembrane leap of pH in chloroplasts in vitro . In the same year, Y. Kagawa & E. Racker ascertained that the synthesis of ATP occurred on the so-called ‘spheres’ referred to as F 1 subunits of the ATP synthase [ 17 ]. Since then the basic contribution of F o F 1 -ATP synthase (ATP synthase) to the OXPHOS became clear.
Developments in the molecular knowledge regarding ATP synthase have been comprehensively addressed in many reviews [ 18 – 20 ]. In summary, we can say that the theory is based on three basic postulates:
- (1) an electron transport chain, providing the energy for H + transfer from one side to other side of the membrane;
- (2) ATP synthase, synthesizing/hydrolysing ATP through H + translocation; and
- (3) impermeability of the inner mitochondrial membrane to ionic species thereby including protons.
The basic requirement for the OXPHOS is a coupling between redox processes, proton translocation and ATP synthesis. Such global coupling can arbitrarily be divided into two distinct phases: a coupling between the oxidation-reductive process and the protonic translocation, referred as ‘RedOx coupling’ or ‘first coupling’ (see figure 1 , left; see also recent review in [ 21 ]) and the coupling between protons accumulated on the p-side of the membrane moving to the n-side through the ATP synthase, which determines the synthesis of ATP, here referred as ‘proton coupling’ or ‘second coupling’ (see right side of figure 1 ). Considerable attention was devoted in the 1980s and 1990s to clarifying the structural–functional details of the respiratory complexes (I, II, III and IV) and ATP synthase (complex V). Important was the study of respiratory complexes organized in supercomplexes [ 22 – 24 ] with the demonstration that the loss of their aggregation leads to an increase in the production of reactive oxygen species [ 24 , 25 ]. The possible participation of complex V to supercomplexes has never been demonstrated [ 23 ]. The study of supercomplexes has also benefited from extraordinary surveys carried out on X-rays [ 26 ].
By contrast, the ‘proton coupling’ or ‘second coupling’ (see figure 1 , right) appears to be the most critical passage of the whole OXPHOS process. Literature reports several experiments performed with reconstructed systems [ 27 , 28 ] (i.e. ATP synthase incorporated into phospholipid vesicles) carried out about fifteen years after Mitchell's hypothesis. Vesicles obtained from the membranes from the purple Halobacterium salinarum synthesized ATP as a result of illumination, and it was thus demonstrated that proton movements through the membrane support the synthesis of ATP. In 1977, N. Sone et al. observed that ATP synthase purified by Thermophilic bacterium , inserted in artificial membranes, was able to synthesize ATP thanks to a transient shift in membrane potential (Δ Ψ ) induced by valinomycin, allowing rapid passage of K + ions across a membrane on the sides of which different salt concentrations were set [ 28 ]. These experiments demonstrated that proton translocation is the crucial step for the ‘proton coupling’ between protonic movement and ATP synthesis. On this general topic, pivotal is the minireview by Junge [ 29 , p. 197], which on one hand highlights the versatility of the ATP synthase, a nano-machine ‘unique in converting electrochemical, mechanical and chemical forms of energy’ and on the other hand points out that there is still much to be understood about the chemical–physical basis of such process.
3. Controversies about the chemiosmotic theory
A long struggle was necessary for the chemiosmotic theory formulated by Mitchell [ 1 ] to be widely accepted. The controversy, central to the history of bioenergetics for more than half a century, appears tackled by more than 200 articles and to have lasted until the most recent years. G. F. Azzone, many years ago (1972), published the manuscript ‘Oxidative phosphorylation, a history of unsuccessful attempts: is it only an experimental problem?’ [ 30 ], which already highlighted the non-convincing parts of the theory, wishing for answers from the fine analysis of the macromolecular structures involved in chemiosmosis.
The harshest criticisms came from J. Prebble, who emphasized the lack of experimental data in support of the theory [ 31 ]. W. Junge effectively described, in his recent review ‘Half a century of molecular bioenergetics’, the chronicle of the dispute, which even took harsh tones [ 32 ]. S. Brown & D. C. Simcook [ 33 ] considered the motivations that have convinced the scientific community to accept the chemiosmotic theory. The authors note that: ‘science shows tremendous resistance to change and it takes extraordinary perseverance to persuade the community’ [ 33 , p. 178].
A controversial issue was the correlation between the membrane potential (Δ Ψ ) and the protonmotive force, often considered equivalent entities [ 34 ]. It was postulated that a Δ Ψ with positive charge on the external p-side of the internal mitochondrial membrane and negative on the n-side in contact with mitochondrial matrix ( figure 1 ) would let protons enter the F o rotor that synthesizes ATP in the matrix thanks to its mechanical connection with the F 1 moiety. Protons would gather across the coupling membrane like chemical ions, creating a driving force for F o F 1 -ATP synthase to synthesize ATP, realizing the ‘proton coupling’. However, the yield in ATP poorly correlates with bulk-to-bulk membrane potential [ 35 , 36 ], questioning the basics of chemiosmotic theory [ 37 ].
The award of the 1978 Nobel Prize for chemistry to Peter Mitchell cooled the dispute, but not definitively. In fact, in 1979, there was a heated confrontation published in Trends in Biochemical Sciences between H. Tedeschi, who disproved the idea that the metabolic activity of mitochondria could contribute to membrane potential, and H. Rottenberg, which instead defended Mitchell's theory [ 38 ]. The original theory provides a protonated ‘delocalized coupling’ (as depicted in figure 1 ), as opposed to a ‘localized coupling’ supported by Williams [ 39 ]. Lee [ 40 ] reported a rigorous chemical/physical experiment in favour of localized coupling, demonstrating furthermore that the thylakoid membrane can be a ‘proton capacitor’. The putative existence of a proton capacitor is a matter of great importance, and later, Saeed & Lee [ 10 ] showed that protons can actually accumulate on the membrane surface even though they never reside in the aqueous phase. Moreover, concerning the experimental verification of the ‘proton coupling’, a recent elegant investigation in HeLa cells, bioengineered with green fluorescent protein as pH indicator inserted in respiratory complex III and in F o moiety of ATP synthase, points to a localized coupling [ 41 ].
A report entitled ‘Proton migration along the membrane surface and retarded surface to bulk transfer’ by Heberle et al . [ 11 ] interestingly reconciles the two visions, providing proof that proton transfer from a proton generator (bacteriorhodopsin) to an acceptor (water-soluble pH indicators) is faster if occurring on the membrane rather than when protons are released in the aqueous bulk. Ferguson [ 12 ] emphasized Heberle et al .'s experiments, concluding that the delocalized coupling and lateral proton transfer (localized coupling) between the proton generator and user occurs very rapidly on the membrane, as compared with the slower and transversal passage through the aqueous bulk. In this context, the recent paper from C. von Ballmoos's group observed that Δ Ψ and ΔpH are equivalent for the coupling with ATP synthase [ 34 ]. A primary role for membrane buffering on proton mobility in general can be hypothesized [ 42 ]. The experimental data [ 43 , 44 ] showing a close thermodynamic correlation between valinomycin-induced Δ Ψ and ATP synthesis in reconstituted systems are very important, but it seems plausible that they induce a transmembrane protonic flow that probably differs from the path in a native environment.
Moreover, the eminent English chemist R. J. Williams clearly rejected the hypothesis of the accumulation of protons from p-side: ‘the p-phase corresponds to the infinitely extended external space. If protons are extruded into this “Pacific Ocean”, they would be diluted and the entropic component of the pmf would be lost’ [ 13 , p. 18]. R. Williams observed that ‘protons in the membrane rather than an osmotic trans-membrane gradient of protons were required to drive ATP formation’, based on a series of considerations that excluded the presence of free protons from p-side [ 39 , p. 123]. An elegant demonstration of Williams's localized coupling hypothesis came in 1976 [ 45 ] by an experiment in which purified ATP synthase was added to the octane–water interface. It was observed that protons accumulate in octane, a Brønsted acid, leading to ATP synthesis by ATP synthase. These data have also been recently confirmed [ 46 ]. Eighteen years later, it was reported that ‘our results suggest that protons can efficiently diffuse along the membrane surface between a source and a sink (for example H+-ATP synthase) without dissipation losses into the aqueous bulk’ [ 11 , p. 379]. From all the cited data it can be concluded that protons (or more likely protonic currents) are confined into the membrane, while proton exit from the membrane is to be considered only as a fallback way of escape, mostly via in vitro reconstituted conditions.
A direct measurement of membrane potential of the mitochondrial inner membrane with microelectrodes was only be accomplished by H. Tedeschi, who showed the existence of a positive inside and negative outside mitochondrial Δ Ψ [ 47 , 48 ]. Such potential (contrary to the canonical one) interestingly coincides with that calculated on the basis of the ionic species present on the membrane sides [ 49 ]. Clearly, knowledge of the entity and especially the sign of this potential is fundamental for understanding the basic functioning of chemiosmosis, as emerges from the already mentioned historical dispute between Tedeschi and Rottenberg [ 38 ].
To measure Δ Ψ , laboratory tests currently use lipophilic fluorescent compounds whose response is considered to be related to Δ Ψ . However, tests conducted with rhodamine have cast doubts on such correlation [ 50 ] since these indicators inhibited the mitochondrial respiration, so they disturb the system. As such compounds dissolve into the membranes, they may reflect the membrane behaviour; in fact they inhibit a membrane intrinsic process (i.e. the OXPHOS), but do not interfere with Δ Ψ . Surely, in vitro , proton passage across membranes can be forced with rapid movements of potassium ions by addition of valinomycin [ 28 , 51 ]. A laboratory procedure using valinomycin and also nigericin has been widely used to create a transient Δ Ψ operating a delocalized coupling linking Δ Ψ and ATP synthesis, but this does not exclude that in the native membranes a localized coupling would operate independently of Δ Ψ .
4. A crucial issue: the membrane permeability to protons
In a previous study (1986), M. Zoratti et al . highlighted the uncertainties of the proton cycle [ 52 ]. In the same year Grzesiek & Dencher [ 53 ] showed that the phospholipid membranes are intrinsically permeable to protons. Data show that phospholipid membranes, normally impermeant to ionic solutes (transversal or permeability coefficients varying between 10 −12 to 10 −14 cm s −1 ), exhibit a significant proton permeability, varying from 10 −3 to 10 −9 cm s −1 . Such variability may be justified by a buffering capacity of the membranes for protons: proton diffusion value could depend on the higher or lower degree of pre-existing protonation. Recently, the proton leak through lipid bilayers was modelled as a concerted mechanism [ 53 – 55 ]. Tepper & Voth [ 54 ] provided a theoretical interpretation of proton permeability, based on the formation of transient membrane spanning aqueous solvent structure. High proton permeability has also been confirmed in liposomes, independently form their phospholipid composition [ 56 ]. It is clear that a high degree of permeability to protons is per se in contrast to the third of the aforementioned basic postulates of the chemiosmotic theory. With regard to the relationship between the membrane and aqueous phases, many observations confirm the existence of a layer of water molecules on the two sides of the membrane, which to some extent isolates it from the aqueous phases present on its two sides [ 57 – 59 ]. In particular, E. Deplazes et al . observed that at the membrane level, H 3 O + forms strong and long-lived hydrogen bonds with the phosphate and carbonyl oxygens in phospholipids [ 60 ].
5. Proton solvation
A central issue is the actual chemical species of the proton: free, or in the form of H 3 O + ? This depends on the phase in which the proton is located. Protons possess peculiar chemical properties, being essentially an atomic nucleus. Free protons do not exist in the aqueous phase, being solvated to H 3 O + , from which the extraction of a proton would be virtually impossible. In fact, in the transition from H 3 O + to free proton a strong energy barrier higher than 500 meV must be overcome. The desolvation barrier [ 61 ] corresponds to the enormous amount of 262 400 Cal mol −1 [ 62 ]. An immense literature exists on the subject [ 40 , 61 , 63 , 64 ]. An interesting report [ 65 ], not sufficiently taken into account, calculated the number of free protons (actually in the form of H 3 O + ) in the volume of a mitochondrion, whose the order of magnitude is femtolitres. Starting from basic physical chemical data (Avogadro number, ionic water product, mathematical pH expression and mitochondrial volume), this study demonstrated that free protons in a mitochondrial periplasmic space are too few (fewer than 10) to support any process dependent on proton translocation in the aqueous bulk across the membrane and absolutely inadequate to support the thousands of ATP synthase molecules present in a mitochondrion. Moreover, the pH value inside the mitochondrion was shown to differ by 0.5 units from what was previously believed [ 66 ]. Indeed, the huge energy associated with proton solvation would have a negative consequence: a free membrane proton would quickly be ‘sucked’ by the near aqueous phase, releasing the huge energy associated with the solvation process, to the detriment of the membrane.
6. Grotthuss mechanism and proton translocation through the membranes
A putative mechanism for proton diffusion was hypothesized more than two centuries ago by von Grotthuss [ 67 ] and is synthetically explained by S. Serowy and colleagues: ‘Proton diffusion according to the Grotthuss mechanism occurs much faster than molecular diffusion because it is uncoupled from the self-diffusion of its mass’ [ 68 , p. 1031]. Protons would not diffuse as a mass, rather as a charge, the latter moving between water molecules or protonable groups of suitable macromolecules of the membranes. The Grotthuss mechanism allows better understanding of the possible ways in which protons or species derived from them move in biological systems. T. E. DeCoursey published a review [ 68 ] which exhaustively analyses proton transfer pathways in water and biological membranes [ 9 ], including Hv1 channels that specifically transfer protons in aqueous phase, therefore actual acidity from one side of the membrane to the other. The mechanism of the voltage gated proton channel Hv1 is not as yet resolved, as emblematically stated by the title of the paper: ‘The voltage-gated proton channel: a riddle, wrapped in a mystery, inside an enigma’ [ 69 ]. Two mechanisms have been proposed for Hv1, as schematically depicted in figure 2 . On the left side of figure 2 is the so-called ‘frozen water’ mechanism, in which the channel traps one or more molecules of water allowing protons to pass through with Grotthuss-style proton hopping, as in the typical case of Gramicidin [ 70 ]. On the right side of figure 2 is a passage of protons with protonation/deprotonation of amino acid side-chains, which would realize the so-called ‘proton wire’ already proposed in the paper by Nagle & Morowitz [ 71 ]. The topic was consolidated successively by Nagle & Tristram-Nagle [ 72 ]. T. E. DeCoursey in a debate recently published in the Journal of Physiology supports the mechanism shown in the right part of figure 2 (i.e. proton wires through the membrane [ 73 ]), while Bennett & Ramsey [ 74 ] support a mechanism of passage through water molecules as schematized on figure 2 , left. Interestingly, both mechanisms are based on the Grotthuss proton movement between (i) water molecules in the case of the water channel (on the left) and (ii) amino acid side chains in the case of protons wires (on the right).

Mechanism for H + transfer through the membrane by H V 1. On the left is the water channel model: the water molecules allow protons to pass through with Grotthuss-style H + hopping. On the right is the proton wire model: a charge migration occurs (through with Grotthuss-style H + hopping) on polar groups of side chains of amino acids of H V 1.
The proton movement in membranes (both biological and artificial) has been analysed by many rigorous chemical/physical studies. The article ‘“Proton holes” in long-range proton transfer reactions in solution and enzymes: a theoretical analysis’ shows that other compounds in addition to water are involved in the ‘proton hopping’ [ 75 ], and is interesting that a quantum-mechanical approach is applied [ 76 ]. The article ‘Grotthuss mechanisms: from proton transport in proton wires to bioprotonic devices’ presents devices such as proton diodes, transistors, memories and transducers, semiconductor electronic devices that use the Grotthuss mechanism [ 77 ].
Notably, Hv1 only allows the passage of protons in the form of hydronium ions, balancing their concentration between two aqueous compartments separated by a membrane. This property does depend on the membrane potential Δ Ψ , similarly to other ion membrane devices abundant in biological membranes (for example the Na + and K + voltage gated channels), acting on the conformation of Hv1. It appears therefore that to carry protons through the membrane there are ad hoc structures that deeply differ from the respiratory complexes, both for their finality and the molecular mechanism. From this and other evidence it can be concluded that the native proton movements in the respiring membranes, when there is no need for acidification of the milieu on one side of a membrane, must take place entirely inside the membrane [ 78 ]. As far as the respiratory complexes are concerned, on the other hand, it is theorized that the proton and membrane potential movements are mutually dependent, in that the pumping of protons would generate the membrane potential, which is impossible for thermodynamic considerations that we will elaborate later on.
Moreover, this comparison between proton movement in support of the OXPHOS and the actual protonic movement in nature sheds light on the fact that when protons are really transferred through a membrane (i) they are never in the form of free protons and (ii) they are subject to the Grotthuss-style proton hopping. Hence the need for chemiosmotic theory to be updated in the light of all this emerges.
7. Inhibition of ATP hydrolysis: an open topic
ATP synthase inherent catalytic properties differentiate it from the vast majority of other enzymes/catalysts. In fact, it transfers energy to the reaction it catalyses, therefore it influences the equilibrium of the reaction while the other enzymes are irrelevant with respect to it. This peculiar property also requires that the activity of ATP synthase be subjected to proper control, as once the coupling with the oxide/reduction systems is lost, the nanomotor itself rapidly hydrolyses ATP, resulting fatal for cell survival. The significant free energy release (Δ G ° = −7300 cal mol −1 ) involved in ATP hydrolysis thus establishes a clear directionality towards the process of hydrolysis itself. It should be emphasized that for most enzymes, the direction of the reaction depends on the concentration of the reactants/substrates, which obviously does not occur for ATP synthase.
To inhibit the reversal of ATP synthase in uncoupled conditions, the action of the inhibitor protein IF1 (UniProtKB: {"type":"entrez-protein","attrs":{"text":"P01096","term_id":"124013","term_text":"P01096"}} P01096 ) is essential, particularly in the brain [ 79 ], which is relatively devoid of energy reserves (glycogen, triglycerides), where anoxia would lead to the tumultuous ATP hydrolysis. IF1 knock down in HeLa cells [ 80 ] equipped with a FRET sensor for measuring the intracellular ATP concentration, surprisingly, did not affect cell viability or mitochondrial morphology, even though the cellular ATP concentration decreased by about a third. Moreover, recently it was reported that deleting the IF1-like ζ subunit from Paracoccus denitrificans has little influence on ATP hydrolysis by ATP synthase [ 81 ], confirming that the inhibition of the hydrolysis of ATP by uncoupled ATP synthase does not depend only on IF1 and that the subject requires further investigations. Also, in bacteria, when the concentration of the ATP reaches the physiological value, the ɛ subunit inhibits ATP synthase by binding to the c ring, and it has been proposed as a target for the design of anti-tuberculosis drugs [ 82 ].
8. Local processes for the coupling inside the respiring membranes
Having established the clear divergence between the pathways of the solvated proton and of the proton alone, detailed molecular structural data on the respiratory complexes able to handle protons (i.e. complex I, III and IV) are now available thanks to the progress of X-ray analysis and of cryo-microscopy [ 2 – 7 ]. Excellent investigations are available on complex I; here for simplicity we only mention the studies from L. Sazanov and collaborators [ 2 , 3 ].
Numerous structural X-ray studies were conducted on complex I from Escherichia coli [ 4 ], Thermus thermophilus [ 2 ] and mammalian ovine ( Ovis aries ) mitochondria [ 5 ], and with cryo-microscopy on complex I from Bos taurus [ 6 , 7 ]. The complexity of the macromolecular aggregation of complex I is impressive: in mammals it is formed by as many as 45 polypeptides and its assembly needs an unknown number of chaperones, so indispensable that the impairment of just one of them (B17.2 L) causes a progressive encephalopathy [ 83 ]. Ogilvie et al. [ 83 , p. 2784] state that ‘results demonstrate that B17.2 L is a bona fide molecular chaperone that is essential for the assembly of complex I and for the normal function of the nervous system.’
We may seek for the proton plausible pathways inside the respiring membrane, even if it is a plasma-membrane. As only five alpha-helices have been found in the complex I structure, in order to allow for proton translocation, it was necessary to postulate the existence of two hemi channels: one from the matrix (n) side to the centre of the respiratory complex, referred here to as the ‘proton entrance hemi channel’, and the other from the centre to the periplasmatic (p) side, here indicated as ‘proton exit hemi channel’. However, only the latter was well identifiable in complex I [ 4 ]. Instead, the entry pathway has not been identified with certainty, so we only can talk about putative pathways labelled with ‘?’ [ 4 ]. Furthermore, it clearly emerges from the X-ray studies that there is an obvious proton tunnelling at the centre of the complex I. Comprehensive studies on the protonic movement inside complex I have been carried out by the Helsinki Bioenergetic Group of M. Wikström, which highlighted uncertainty margins on the stoichiometry of protonic extrusion that appear closer to 3 H + /2e − [ 84 ] instead to the classic 4 H + /2e − . Also, in the review of Verkhovskaya & Bloch [ 85 ] (of Helsinki Bioenergetic Group), four mechanisms for proton translocation are proposed and the ‘proton entrance half channel’ is not identified with certainty, while the ‘proton exit half channel’ is clearly identifiable. Emblematically, on the website of the Helsinki Bioenergetic Group it is written ( www.biocenter.helsinki.fi/bi/hbg/cl/complex_I_main.html ) that ‘the mechanism of proton transfer in complex I remains completely enigmatic’.
As far as complex IV (cytochrome c oxidase) is concerned, the proton translocation of has been studied in depth [ 86 ]. In their recent review, M. Wikström & V. Sharma talk of an ‘anniversary’: ‘Proton pumping by cytochrome c oxidase—A 40 year anniversary’ [ 87 ]. Among the many works cited in this review the Chemical Review article of the M. Wikström group stands out [ 8 ]. It goes into the details of the possible molecular processes carried out by complex IV [ 8 ], thereby including proton translocation. The topic is complex, and more putative pathway of protons are well developed in the review, which we cannot here detail here. For the ‘proton entrance half channel’, for each molecule of oxygen reduced to water they need 4 H + , and it is hypothesized that another 4 H + (for a total of 8) enter through this channel. It seems unlikely that there exists equivalence between protons that exist as particles and link to water molecules and protons that should move as a charge, according to the Grotthuss mechanism.
9. Proposal for a localized complex I-ATP synthase coupling
Taking into account what is reported above, it is possible to trace a plausible proton pathway within the respiring membrane. In 2006 a direct proton transfer was proposed to couple the respiratory pathway of complexes I, III, IV with ATP synthase [ 88 ]. However, in a recent review [ 21 ] the concept of transmembrane proton motor force to move the ATP synthase is reinforced, although it is noted that many aspects of coupling are not yet clarified. Clear-cut consideration was proposed some years ago (1991) by Akeson & Deamer [ 89 ] about speed of proton translocation through putative proton channel as a limiting step for ATP synthesis by ATP synthase. For the sake of simplicity, we only examine the coupling between the respiratory complex I and the ATP synthase. The existence of a proton pathway at the centre of complex I is quite clear, and we can hypothesize that the protons are sent to the well-identified ‘exit half channel’, as shown in figure 3 . The proton donor at the centre of complex I has already been tentatively identified in the phospholipid cardiolipin (CL) [ 78 ], essential for OXPHOS. However, for example, phosphatidylethanolamine (PE) can effectively sustain the functioning of OXPHOS [ 90 ]. Data from C. von Ballmoos's group [ 91 , 92 ] unconventionally clarify such a role of phospholipids. In fact, pure phosphatidylcholine (PC) is excellent for the coupling, increasing when the membrane is formed by PC + PE. By contrast, coupling is dramatically inhibited if the membrane is formed by PC + CL, incredibly diverging from the traditional role assigned to CL. This research is also important because experiments are performed with a reconstructed system, more adherent to the ‘H + /ATP coupling’ or ‘second coupling’ ( figure 1 ). Here, the driving force that feeds ATP synthase was not the rapid transfer of K + generated by valinomycin, a widely used method, but the bo 3 oxidase of Escherichia coli , analogous to the respiratory complex I of vertebrates. It emerges that all membrane phospholipids can act as mobile proton transporters, hindering proton relocation in the near-aqueous medium, so that these are never free. The exergonic process of proton solvation would release an impressive amount of energy (262 400 Cal mol −1 ) [ 62 ], which, if not transferred on a generic acceptor, would generate heat devastating not only the membrane but also the cell integrity. Complex I would transfer protons to the p side of the membrane, but these would not be dispersed in the aqueous bulk. In fact it is well documented that there exists a barrier of water molecules attached to the membrane, determining the lateral displacement of protons on the phosphate heads of phospholipids [ 68 , 93 , 94 ] to meet a sink that is the ATP synthase subunit which, through a proton wire [ 95 , 96 ], would lead the proton to the rotor ( figure 3 ), accommodating on the central glutamic or aspartic residue (depending on the species) of the c subunit, which in variable numbers from 8 to 15 make up the rotor F o [ 97 ]. As F o rotates it is conceivable that the proton returns to the centre of complex I, a highly hydrophobic environment, thus closing the protonic circuit. This last passage, from the rotor exit to the centre of the respiratory complex, appears plausible for the operativity of the Brownian motion (diffusion) of particles in a highly anisotropic environment that can occur efficiently [ 45 , 98 ] and for a theoretically possible direct passage of the proton exiting the subunit at the entrance to the complexes. Moreover the investigations of the C. von Ballmoos group have produced exhaustive studies with reconstructed systems [ 91 , 92 , 99 ], in which a direct transfer of proton on the p-side of the membrane from complex I to ATP synthase is evident. Their recent paper also demonstrates that proximity to the membrane between the respiratory complex and ATP synthase is required for the ‘proton coupling’ [ 92 ]. However, the fact remains that the last part of the protonic circuitry (i.e. from F o moiety of ATP synthase to respiratory complexes) is only hypothetical.

A possible H + circuit inside respiring membrane. The phosphate groups of phospholipids on both sides of the membrane are shown by brown ellipsoids. The image proposes that the H + (red dotted line) are transferred to the Glu 58 (E58) at the centre of subunit c through subunit a of ATP synthase by proton tunnelling. H + would flow from the periplasmic side, always bound to phospholipid heads. This can be arranged in each layer of the membrane.
In this controversial scenario, a decisive contribution is undoubtedly the sophisticated bioengineering experiment that labelled both complex IV and ATP synthase with proteins of the GFP family, to experimentally observe a local ΔpH triggered by the respiratory substrate galactose [ 41 ]. Observations were conducted in cultured HeLa cells. The authors concluded [ 41 , p. 1]: ‘the observed lateral variation in the proton-motive force necessitates a modification to Peter Mitchell's chemiosmotic proposal’. In other words, the experimentally proven lateral proton motive force is in line with the hypothesis of localized coupling.
10. Extramitochondrial oxidative phosphorylation
The chemiosmotic theory [ 1 ] as it was formulated envisions a process that can only take place in organelles possessing double membrane systems forming closed compartments to entrap protons, such as mitochondrial cristae, bacteria and thylakoids (here, for the sake of simplicity, we have considered the mitochondrial inner membrane). However, in the last years, several authors have described a functional expression of OXPHOS machinery in cellular districts devoid of mitochondria [ 100 , 101 ], suggesting a possible build-up of a transversal proton gradient across the membrane, out of mitochondria. In particular, an extramitochondrial OXPHOS has been described in rod outer segment (OS) discs [ 102 – 105 ], myelin sheath [ 79 , 106 – 110 ], cell plasma membrane [ 111 – 119 ], platelets [ 120 ], and extracellular vesicles shedding from cells such as exosomes and microvesicles [ 121 , 122 ], which seem to carry an unsuspected metabolic signature [ 121 , 123 ]. Notably, since the plasma membrane potential is positive on the outside and negative on the inside, it would favour ATP hydrolysis rather than its synthesis. The extramitochondrial ATP synthesis is in line with the postulated independence of ATP synthase activity from the membrane potential, and therefore it is plausible that this synthesis of ATP depends on the proton intramembranous coupling.
11. Conclusion
The impressive amount of experimental data cited appears globally in contrast with the above-cited three assumptions underlying the chemiosmotic theory. First of all, it excludes that the protons can accumulate on the coupling membrane surface, whose high permeability would dissipate, since it would correspond to an extreme acidity incompatible with any vital process. Second, since proton diffusion does occur, it is clear that the membrane potential is irrelevant to proton translocation. Third, it can be excluded that the respiratory complexes operate as transmembrane proton transfer from the aqueous bulk, where the proton would exist as hydroxonium ion. This appears to rule out the actual possibility that ATP synthase can overcome the energy barrier, higher than 500 meV [ 61 ], required to extract the proton from water, leaving space for the localized coupling hypothesis [ 63 , 64 ], as a dehydration–hydration reaction from hydroxonium ion to free protons would require 262 400 Cal mol −1 [ 62 ]. Which are the actual processes acting on the ATP synthase? There is no certainty, as highlighted by the emblematic title of an article by J. Walker: ‘The ATP synthase: the understood, the uncertain and the unknown’ [ 124 ].
Indeed, today a constellation of clues leads us to hypothesize the existence of protonic currents internal to the membrane, with the formation of possible circuits travelled by the positive elementary charge, thus realizing a localized coupling that excludes an osmotic nature of the process. To trace this circuit, at least for the possible coupling between respiratory complex I and ATP synthase, it appears realistic that complex I may transfer protons from its central part to the periplasmatic side, allowing them to travel on the membrane surface thanks to the heads of phospholipids [ 93 , 94 ] finally tunnelling inside the a subunit of ATP synthase [ 95 , 125 ] ( figure 3 ). Moreover, several studies show that the membrane is isolated from the aqueous bulk thanks to a layer of water molecules on both sides of the membrane, which consolidates the idea that the membrane is radically distinct and isolated from the liquid phase [ 59 ]. Considering the two isolated phases, the only way evolution could pursue to link proton movement to ATP synthesis was a nanomachine connecting the proton movement inside the membrane to the deformation mechanics of the F 1 sphere immersed in the aqueous phase. We can reasonably assume that the proton movement inside the membranes occurs as a charge, according to the proton-hopping Grotthuss mechanism, with the establishment of ‘protonic currents’ inside the membrane [ 126 ]. In the non-biological field we find a remarkable adherence to this theory for the development of protonic devices (as proton diodes, transistors, memories and transducers) [ 77 ]. It is surprising that the biological and the physical–chemical areas have ignored each other. Since in the history of biology the application of physical–chemical methodologies has led to dramatic advances in biology (we may remind the reader that the resolution of DNA structure [ 127 ] was obtained with the fundamental application of X-ray crystallography, developed in 1913 by Williams Henry Bragg and his son Lawrence for the study of inorganic crystals), it is desirable that this fusion of knowledge can be realized in the years to come.
Furthermore, the classical mechanical approach cannot be used to approach these currents. In fact, any time there is a movement of charge bound to a mass, the dualism that cannot be assessed by classical mechanics, instead quantum mechanics must be applied, and in this perspective lay the promising recent quantum-mechanical approaches by Riccardi et al. [ 76 ] and Ivontsin et al . [ 125 ]. There is a need to take into account the Heisenberg indeterminacy principle as Philip Hunter already highlighted in the title of a 2006 paper: ‘A quantum leap in biology: one inscrutable field helps another, as quantum physics unravels consciousness’ [ 128 ].
Supplementary Material
Acknowledgements.
The authors are very grateful to Professor Giorgio Lenaz, Bologna University (Italy), for the critical reading of the manuscript and for the valuable comments and discussions, and Professor Matthew Stephen Hodgart, University of Surrey, for help in revising the manuscript language.
Data accessibility
Competing interests.
We declare we have no competing interests.
We received no funding for this study.
Want to create or adapt books like this? Learn more about how Pressbooks supports open publishing practices.
9.4 Coupling and Repulsion (cis and trans) Configuration
Just by looking at an organism that is heterozygous at two loci, you cannot tell how the mutant and wild type alleles are arranged. Both mutant alleles could be on one homologous chromosome, and both wild type alleles could be on the other (e.g., a – b – / A + B + ). This is known as a coupling (or cis) configuration . When one wild type allele and one mutant allele are on one homologous chromosome, and the opposite is on the other, this is known as a repulsion (or trans) configuration (e.g., A + b – / a – B + ). The way to determine the orientation is to look at the parents (or P generation) of that cross if you know the genotypes of them. If the parents are homozygous for both genes, and one shows both dominant phenotypes and the other shows both recessive phenotypes, then you know that the individual you are looking at is in coupling configuration. If one parent has one dominant and one recessive phenotype, and the other has the opposite, then you know the individual is in repulsion configuration.
The following video, Genetics! coupling (cis) vs Repulsion (trans) , by Medaphysics Repository (2015) on YouTube, discusses the difference between cis and trans genes.
The video, Coupling vs Repulsion, by Genetics Rocks (2019) on YouTube, looks at a worked example involving observed frequencies in a text cross and genes in coupling/repulsion.
Media Attributions
- Figure 9.4.1 Original by Deyholos (2017), CC BY-NC 3.0 , Open Genetics Lectures
Deyholos, M. (2017). Figure 5. Alleles in coupling configuration…[digital image]. In Locke, J., Harrington, M., Canham, L. and Min Ku Kang (Eds.), Open Genetics Lectures, Fall 2017 (Chapter 18, p. 4). Dataverse/ BCcampus. http://solr.bccampus.ca:8001/bcc/file/7a7b00f9-fb56-4c49-81a9-cfa3ad80e6d8/1/OpenGeneticsLectures_Fall2017.pdf
Genetics Rocks. (2019, September 14). Coupling vs repulsion (video file). YouTube. https://www.youtube.com/watch?v=llNZP1Wmgok
Medaphysics Repository. (2015, February 24). Genetics! coupling (cis) vs Repulsion (trans) (video file). YouTube. https://www.youtube.com/watch?v=4y5vjhMq6iY
Long Description
- Figure 9.4.1 Two cells showing alleles in either the coupling or cis configuration (whereby both mutant alleles are present on one homologous chromosome, and both wild type alleles are present on the other) or the repulsion or the trans configuration (whereby one wild type allele and one mutant allele are on one homologous chromosome, and the opposite is on the other). [Back to Figure 9.4.1 ]
Introduction to Genetics Copyright © 2023 by Natasha Ramroop Singh, Thompson Rivers University is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License , except where otherwise noted.
Share This Book
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Review Article
- Published: January 2008
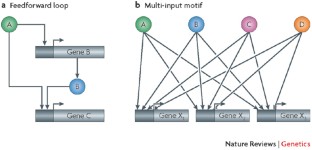
Coupling and coordination in gene expression processes: a systems biology view
- Suzanne Komili 1 , 2 &
- Pamela A. Silver 1
Nature Reviews Genetics volume 9 , pages 38–48 ( 2008 ) Cite this article
4306 Accesses
153 Citations
1 Altmetric
Metrics details
Traditionally, the different stages of gene expression were considered to be separate processes that operated independently. Biochemical and genetic studies have instead revealed high levels of coupling between the different stages. In recent years, genome-wide and systems-level analyses have greatly extended our understanding of coupling in gene expression, by both revealing the effect that certain forms of coupling have on a more global scale, and unveiling novel forms of coupling that had not been identified using more traditional techniques.
Genomic analyses of the binding of transcription factors and chromatin remodellers with DNA have revealed extensive coordination and coupling between these factors, forming regulatory networks that control how many genes are expressed.
Genome-wide analysis of associations between the nuclear pore and nuclear lamina revealed extensive coupling between the transcription level of a given gene and its nuclear localization. In particular, different subcomplexes within the nuclear pore associate with specific types of genetic loci.
Genomic analyses of mRNA processing have shown that splicing commitment occurs co-transcriptionally, but that in yeast the splicing completes post-transcriptionally, and that the spliceosome can regulate expression of specific types of mRNAs by increasing or decreasing their splicing efficiencies.
Microarray analyses have also shown that the co-transcriptional recruitment of mRNA export factors results in functional specificities, in which different factors transport specific types of mRNAs; the combination of whole-genome screens and array analyses also demonstrated an important role for the exosome in mRNA export.
Chromatin immunoprecipitation coupled with microarray (ChIP–chip) analyses in higher eukaryotes provide support for a dynamic messenger ribonucleoprotein (mRNP) model, in which the composition of the mRNP associated with the nascent transcript changes along the length of the transcript.
Whole-genome analysis of the associations between proteasomal components and chromatin revealed a widespread role for the proteasome in transcriptional activation and enrichment of binding of the proteasome at ribosomal protein genes. This coupling between the protein synthesis and degradation machineries could allow feedback that globally reduces expression levels when the proteasome is compromised.
Network analysis of the genomic associations of many factors involved in gene expression revealed novel connections between the different levels of gene expression. In particular, factors involved in nuclear transport, general transcription factors, RNA-processing factors and nucleosome remodellers were found to exhibit highly similar binding profiles and to have a large number of neighbours, allowing them to exert global influences on many genes at once.
Genome-scale analyses have allowed us to progress beyond studying gene expression at the level of individual components of a given process by providing global information about functional connections between genes, mRNAs and their regulatory proteins. Such analyses have greatly increased our understanding of the interplay between different events in gene regulation and have highlighted previously unappreciated functional connections, including coupling between nuclear and cytoplasmic processes. Genome-wide approaches have also revealed extensive coordination within regulatory levels, such as the organization of transcription factors into regulatory motifs. Overall, these studies enhance our understanding of how the many components of the eukaryotic cell function as a system to allow both coordination and versatility in gene expression.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
176,64 € per year
only 14,72 € per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others

A unified framework for integrative study of heterogeneous gene regulatory mechanisms
Qin Cao, Zhenghao Zhang, … Kevin Y. Yip

Nuclear compartmentalization as a mechanism of quantitative control of gene expression
Prashant Bhat, Drew Honson & Mitchell Guttman

The relationship between genome structure and function
A. Marieke Oudelaar & Douglas R. Higgs
Maniatis, T. & Reed, R. An extensive network of coupling among gene expression machines. Nature 416 , 499–506 (2002).
Article CAS PubMed Google Scholar
Soller, M. Pre-messenger RNA processing and its regulation: a genomic perspective. Cell. Mol. Life Sci. 63 , 796–819 (2006).
Li, B., Carey, M. & Workman, J. The role of chromatin during transcription. Cell 128 , 707–719 (2007).
Squazzo, S. et al. Suz12 binds to silenced regions of the genome in a cell-type-specific manner. Genome Res. 16 , 890–900 (2006).
Article CAS PubMed PubMed Central Google Scholar
Mikkelsen, T. et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448 , 553–560 (2007).
Funayama, R. & Ishikawa, F. Cellular senescence and chromatin structure. Chromosoma 116 , 431–440 (2007).
Article PubMed Google Scholar
Polo, S. & Almouzni, G. Histone metabolic pathways and chromatin assembly factors as proliferation markers. Cancer Lett. 220 , 1–9 (2005).
Lee, T. & Young, R. Transcription of eukaryotic protein-coding genes. Annu. Rev. Genet. 34 , 77–137 (2000).
Narlikar, G., Fan, H. & Kingston, R. Cooperation between complexes that regulate chromatin structure and transcription. Cell 108 , 475–487 (2002).
Lee, T. et al. Transcriptional regulatory networks in Saccharomyces cerevisiae . Science 298 , 799–804 (2002). Reference 10 uses ChIP–chip analysis of all transcription factors in yeast to provide a view of regulatory mechanisms among, and feedback between, all transcription factors.
CAS PubMed Google Scholar
Sandmann, T. et al. A core transcriptional network for early mesoderm development in Drosophila melanogaster . Genes Dev. 21 , 436–449 (2007).
Cao, Y. et al. Global and gene-specific analyses show distinct roles for MYOD and MYOG at a common set of promoters. EMBO J. 25 , 502–511 (2006).
Carroll, J. et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 122 , 33–43 (2005).
Scacheri, P. et al. Genome-wide analysis of menin binding provides insights into MEN1 tumorigenesis. PLoS Genet. 2 , e51 (2006).
Lanctot, C., Cheutin, T., Cremer, M., Cavalli, G. & Cremer, T. Dynamic genome architecture in the nuclear space: regulation of gene expression in three dimensions. Nature Rev. Genet. 8 , 104–115 (2007).
Akhtar, A. & Gasser, S. M. The nuclear envelope and transcriptional control. Nature Rev. Genet. 8 , 507–517 (2007).
Cabal, G. et al. SAGA interacting factors confine sub-diffusion of transcribed genes to the nuclear envelope. Nature 441 , 770–773 (2006).
Taddei, A. et al. Nuclear pore association confers optimal expression levels for an inducible yeast gene. Nature 441 , 774–778 (2006).
Dieppois, G., Iglesias, N. & Stutz, F. Cotranscriptional recruitment to the mRNA export receptor Mex67p contributes to nuclear pore anchoring of activated genes. Mol. Cell Biol. 26 , 7858–7870 (2006).
Brickner, J. & Walter, P. Gene recruitment of the activated INO1 locus to the nuclear membrane. PLoS Biol. 2 , e342 (2004).
Casolari, J. et al. Genome-wide localization of the nuclear transport machinery couples transcriptional status and nuclear organization. Cell 117 , 427–439 (2004). Reference 21 provides the first global view of gene associations with the nuclear pore. Using ChIP–chip analysis it shows that specific subcomplexes within the nuclear pore associate with different types of genes.
Casolari, J., Brown, C., Drubin, D., Rando, O. & Silver, P. Developmentally induced changes in transcriptional program alter spatial organization across chromosomes. Genes Dev. 19 , 1188–1198 (2005).
Pickersgill, H. et al. Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nature Genet. 38 , 1005–1014 (2006).
Mendjan, S. et al. Nuclear pore components are involved in the transcriptional regulation of dosage compensation in Drosophila . Mol. Cell 21 , 811–823 (2006).
Alekseyenko, A. A., Larschan, E., Lai, W. R., Park, P. J. & Kuroda, M. I. High-resolution ChIP–chip analysis reveals that the Drosophila MSL complex selectively identifies active genes on the male X chromosome. Genes Dev. 20 , 848–857 (2006).
Gilfillan, G. D. et al. Chromosome-wide gene-specific targeting of the Drosophila dosage compensation complex. Genes Dev. 20 , 858–870 (2006).
Legube, G., McWeeney, S. K., Lercher, M. J. & Akhtar, A. X-chromosome-wide profiling of MSL-1 distribution and dosage compensation in Drosophila . Genes Dev. 20 , 871–883 (2006).
Cohen, B., Mitra, R., Hughes, J. & Church, G. A computational analysis of whole-genome expression data reveals chromosomal domains of gene expression. Nature Genet. 26 , 183–186 (2000).
Keene, J. RNA regulons: coordination of post-transcriptional events. Nature Rev. Genet. 8 , 533–543 (2007).
Beyer, A. L. & Osheim, Y. N. Splice site selection, rate of splicing, and alternative splicing on nascent transcripts. Genes Dev. 2 , 754–765 (1988).
Lacadie, S. & Rosbash, M. Cotranscriptional spliceosome assembly dynamics and the role of U1 snRNA:5′ss base pairing in yeast. Mol. Cell 19 , 65–75 (2005).
Gornemann, J., Kotovic, K. M., Hujer, K. & Neugebauer, K. M. Cotranscriptional spliceosome assembly occurs in a stepwise fashion and requires the cap binding complex. Mol. Cell 19 , 53–63 (2005).
Moore, M., Schwartzfarb, E., Silver, P. & Yu, M. Differential recruitment of the splicing machinery during transcription predicts genome-wide patterns of mRNA splicing. Mol. Cell 24 , 903–915 (2006).
Tardiff, D., Lacadie, S. & Rosbash, M. A genome-wide analysis indicates that yeast pre-mRNA splicing is predominantly posttranscriptional. Mol. Cell 24 , 917–929 (2006). References 33 and 34 use ChIP–chip analysis to demonstrate that the majority of splicing in yeast occurs post-transcriptionally, although the initiation factors are recruited during transcription.
Listerman, I., Sapra, A. & Neugebauer, K. Cotranscriptional coupling of splicing factor recruitment and precursor messenger RNA splicing in mammalian cells. Nature Struct. Mol. Biol. 13 , 815–822 (2006).
Article CAS Google Scholar
Tennyson, C., Klamut, H. & Worton, R. The human dystrophin gene requires 16 hours to be transcribed and is cotranscriptionally spliced. Nature Genet. 9 , 184–190 (1995).
Pleiss, J. A., Whitworth, G. B., Bergkessel, M. & Guthrie, C. Transcript specificity in yeast pre-mRNA splicing revealed by mutations in core spliceosomal components. PLoS Biol. 5 , e90 (2007).
Pleiss, J. A., Whitworth, G. B., Bergkessel, M. & Guthrie, C. Rapid, transcript-specific changes in splicing in response to environmental stress. Mol. Cell 27 , 928–937 (2007). References 37 and 38 use custom–designed microarrays to analyse the splicing efficiencies of individual mRNAs in response to mutations in core components of the spliceosome and to certain stress conditions. This analysis demonstrates regulation of specific types of genes by the splicing machinery in response to cellular stresses.
Gama-Carvalho, M., Barbosa-Morais, N., Brodsky, A., Silver, P. & Carmo-Fonseca, M. Genome-wide identification of functionally distinct subsets of cellular mRNAs associated with two nucleocytoplasmic-shuttling mammalian splicing factors. Genome Biol. 7 , R113 (2006).
Ule, J. et al. CLIP identifies Nova-regulated RNA networks in the brain. Science 302 , 1212–1215 (2003).
Ule, J. et al. Nova regulates brain-specific splicing to shape the synapse. Nature Genet. 37 , 844–852 (2005).
Jensen, K. B. et al. Nova-1 regulates neuron-specific alternative splicing and is essential for neuronal viability. Neuron 25 , 359–371 (2000).
Lei, E., Krebber, H. & Silver, P. Messenger RNAs are recruited for nuclear export during transcription. Genes Dev. 15 , 1771–1782 (2001).
Lei, E. & Silver, P. Intron status and 3′-end formation control cotranscriptional export of mRNA. Genes Dev. 16 , 2761–2766 (2002).
Kim Guisbert, K., Duncan, K., Li, H. & Guthrie, C. Functional specificity of shuttling hnRNPs revealed by genome-wide analysis of their RNA binding profiles. RNA 11 , 383–393 (2005).
Hieronymus, H. & Silver, P. Genome-wide analysis of RNA–protein interactions illustrates specificity of the mRNA export machinery. Nature Genet. 33 , 155–161 (2003).
Herold, A., Teixeira, L. & Izaurralde, E. Genome-wide analysis of nuclear mRNA export pathways in Drosophila . EMBO J. 22 , 2472–2483 (2003).
Swinburne, I., Meyer, C., Liu, X., Silver, P. & Brodsky, A. Genomic localization of RNA binding proteins reveals links between pre-mRNA processing and transcription. Genome Res. 16 , 912–921 (2006).
Buttner, K., Wenig, K. & Hopfner, K. P. The exosome: a macromolecular cage for controlled RNA degradation. Mol. Microbiol. 61 , 1372–1379 (2006).
Andrulis, E. et al. The RNA processing exosome is linked to elongating RNA polymerase II in Drosophila . Nature 420 , 837–841 (2002).
West, S., Gromak, N., Norbury, C. & Proudfoot, N. Adenylation and exosome-mediated degradation of cotranscriptionally cleaved pre-messenger RNA in human cells. Mol. Cell 21 , 437–443 (2006).
Brodsky, A. et al. Genomic mapping of RNA polymerase II reveals sites of co-transcriptional regulation in human cells. Genome Biol. 6 , R64 (2005).
Howe, K., Kane, C. & Ares, M. Perturbation of transcription elongation influences the fidelity of internal exon inclusion in Saccharomyces cerevisiae . RNA 9 , 993–1006 (2003).
Gromak, N., West, S. & Proudfoot, N. Pause sites promote transcriptional termination of mammalian RNA polymerase II. Mol. Cell Biol. 26 , 3986–3996 (2006).
Hieronymus, H., Yu, M. & Silver, P. Genome-wide mRNA surveillance is coupled to mRNA export. Genes Dev. 18 , 2652–2662 (2004).
Hilleren, P., McCarthy, T., Rosbash, M., Parker, R. & Jensen, T. Quality control of mRNA 3′-end processing is linked to the nuclear exosome. Nature 413 , 538–542 (2001).
Galy, V. et al. Nuclear retention of unspliced mRNAs in yeast is mediated by perinuclear Mlp1. Cell 116 , 63–73 (2004).
Yu, M. et al. Arginine methyltransferase affects interactions and recruitment of mRNA processing and export factors. Genes Dev. 18 , 2024–2035 (2004).
Wolffe, A. & Meric, F. Coupling transcription to translation: a novel site for the regulation of eukaryotic gene expression. Int. J. Biochem. Cell Biol. 28 , 247–257 (1996).
Belostotsky, D. & Rose, A. Plant gene expression in the age of systems biology: integrating transcriptional and post-transcriptional events. Trends Plant Sci. 10 , 347–353 (2005).
Nott, A., Le Hir, H. & Moore, M. Splicing enhances translation in mammalian cells: an additional function of the exon junction complex. Genes Dev. 18 , 210–222 (2004).
Wiegand, H., Lu, S. & Cullen, B. Exon junction complexes mediate the enhancing effect of splicing on mRNA expression. Proc. Natl Acad. Sci. USA 100 , 11327–11332 (2003).
Windgassen, M. et al. Yeast shuttling SR proteins Npl3p, Gbp2p, and Hrb1p are part of the translating mRNPs, and Npl3p can function as a translational repressor. Mol. Cell Biol. 24 , 10479–10491 (2004).
Sanford, J. R., Gray, N. K., Beckmann, K. & Caceres, J. F. A novel role for shuttling SR proteins in mRNA translation. Genes Dev. 18 , 755–768 (2004).
Lemaire, R. et al. Stability of a PKCI-1-related mRNA is controlled by the splicing factor ASF–SF2: a novel function for SR proteins. Genes Dev. 16 , 594–607 (2002).
Hachet, O. & Ephrussi, A. Splicing of Oskar RNA in the nucleus is coupled to its cytoplasmic localization. Nature 428 , 959–963 (2004).
Oleynikov, Y. & Singer, R. Real-time visualization of ZBP1 association with β-actin mRNA during transcription and localization. Curr. Biol. 13 , 199–207 (2003).
Colón-Ramos, D. et al. Asymmetric distribution of nuclear pore complexes and the cytoplasmic localization of β2-tubulin mRNA in Chlamydomonas reinhardtii . Dev. Cell 4 , 941–952 (2003).
Brown, V. et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 107 , 477–487 (2001).
Takizawa, P., DeRisi, J., Wilhelm, J. & Vale, R. Plasma membrane compartmentalization in yeast by messenger RNA transport and a septin diffusion barrier. Science 290 , 341–344 (2000).
Tenenbaum, S., Carson, C., Lager, P. & Keene, J. Identifying mRNA subsets in messenger ribonucleoprotein complexes by using cDNA arrays. Proc. Natl Acad. Sci. USA 97 , 14085–10490 (2000).
Baumeister, W., Walz, J., Zühl, F. & Seemüller, E. The proteasome: paradigm of a self-compartmentalizing protease. Cell 92 , 367–380 (1998).
Glickman, M., Rubin, D., Fried, V. & Finley, D. The regulatory particle of the Saccharomyces cerevisiae proteasome. Mol. Cell Biol. 18 , 3149–3162 (1998).
Schmidt, M., Hanna, J., Elsasser, S. & Finley, D. Proteasome-associated proteins: regulation of a proteolytic machine. Biol. Chem. 386 , 725–737 (2005).
Muratani, M. & Tansey, W. How the ubiquitin-proteasome system controls transcription. Nature Rev. Mol. Cell Biol. 4 , 192–201 (2003).
Sun, L., Johnston, S. & Kodadek, T. Physical association of the APIS complex and general transcription factors. Biochem. Biophys. Res. Commun. 296 , 991–999 (2002).
Ferdous, A., Gonzalez, F., Sun, L., Kodadek, T. & Johnston, S. The 19S regulatory particle of the proteasome is required for efficient transcription elongation by RNA polymerase II. Mol. Cell 7 , 981–991 (2001).
Gonzalez, F., Delahodde, A., Kodadek, T. & Johnston, S. Recruitment of a 19S proteasome subcomplex to an activated promoter. Science 296 , 548–550 (2002).
Sulahian, R., Sikder, D., Johnston, S. & Kodadek, T. The proteasomal ATPase complex is required for stress-induced transcription in yeast. Nucleic Acids Res. 34 , 1351–1357 (2006).
Gillette, T., Gonzalez, F., Delahodde, A., Johnston, S. & Kodadek, T. Physical and functional association of RNA polymerase II and the proteasome. Proc. Natl Acad. Sci. USA 101 , 5904–5909 (2004).
Ferdous, A., Kodadek, T. & Johnston, S. A nonproteolytic function of the 19S regulatory subunit of the 26S proteasome is required for efficient activated transcription by human RNA polymerase II. Biochemistry 41 , 12798–12805 (2002).
Lipford, J. & Deshaies, R. Diverse roles for ubiquitin-dependent proteolysis in transcriptional activation. Nature Cell Biol. 5 , 845–850 (2003).
Morris, M. et al. Cks1-dependent proteasome recruitment and activation of CDC20 transcription in budding yeast. Nature 423 , 1009–1013 (2003).
Nalley, K., Johnston, S. & Kodadek, T. Proteolytic turnover of the Gal4 transcription factor is not required for function in vivo . Nature 442 , 1054–1057 (2006).
Lipford, J., Smith, G., Chi, Y. & Deshaies, R. A putative stimulatory role for activator turnover in gene expression. Nature 438 , 113–116 (2005).
Nawaz, Z. & O'Malley, B. Urban renewal in the nucleus: is protein turnover by proteasomes absolutely required for nuclear receptor-regulated transcription? Mol. Endocrinol. 18 , 493–499 (2004).
Reid, G. et al. Cyclic, proteasome-mediated turnover of unliganded and liganded ERα on responsive promoters is an integral feature of estrogen signaling. Mol. Cell 11 , 695–707 (2003).
Ferdous, A. et al. The role of the proteasomal ATPases and activator monoubiquitylation in regulating Gal4 binding to promoters. Genes Dev. 21 , 112–123 (2007).
Auld, K., Brown, C., Casolari, J., Komili, S. & Silver, P. Genomic association of the proteasome demonstrates overlapping gene regulatory activity with transcription factor substrates. Mol. Cell 21 , 861–871 (2006).
Sikder, D., Johnston, S. & Kodadek, T. Widespread, but non-identical, association of proteasomal 19 and 20 S. proteins with yeast chromatin. J. Biol. Chem. 281 , 27346–27355 (2006). References 89 and 90 use ChIP–chip analysis of different proteasomal subunits to demonstrate a widespread role for the proteasome in gene activation, as well as regulation of ribosomal protein genes by the binding of proteasomal subunits.
Dembla-Rajpal, N., Seipelt, R., Wang, Q. & Rymond, B. Proteasome inhibition alters the transcription of multiple yeast genes. Biochim. Biophys. Acta 1680 , 34–45 (2004).
Fatica, A., Oeffinger, M., Tollervey, D. & Bozzoni, I. Cic1p–Nsa3p is required for synthesis and nuclear export of 60S ribosomal subunits. RNA 9 , 1431–1436 (2003).
Fleming, J. et al. Complementary whole-genome technologies reveal the cellular response to proteasome inhibition by PS-341. Proc. Natl Acad. Sci. USA 99 , 1461–1466 (2002).
Tsankov, A. et al. Communication between levels of transcriptional control improves robustness and adaptivity. Mol. Syst. Biol. 2 , 65 (2006). Reference 94 describes a network analysis of many different ChIP–chip datasets in yeast and identifies novel forms of coupling between different levels of gene regulation.
Drubin, D., Garakani, A. & Silver, P. Motion as a phenotype: the use of live-cell imaging and machine visual screening to characterize transcription-dependent chromosome dynamics. BMC Cell Biol. 7 , 19 (2006).
Luthra, R. et al. Actively transcribed GAL genes can be physically linked to the nuclear pore by the SAGA chromatin modifying complex. J. Biol. Chem. 282 , 3042–3049 (2007).
Lee, D. et al. The proteasome regulatory particle alters the SAGA coactivator to enhance its interactions with transcriptional activators. Cell 123 , 423–436 (2005).
Zanetti, M., Chang, I., Gong, F., Galbraith, D. & Bailey-Serres, J. Immunopurification of polyribosomal complexes of Arabidopsis for global analysis of gene expression. Plant Physiol. 138 , 624–635 (2005).
Arava, Y. et al. Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiae . Proc. Natl Acad. Sci. USA 100 , 3889–3894 (2003).
MacKay, V. L. et al. Gene expression analyzed by high-resolution state array analysis and quantitative proteomics: response of yeast to mating pheromone. Mol. Cell Proteomics 3 , 478–489 (2004).
Dekker, J., Rippe, K., Dekker, M. & Kleckner, N. Capturing chromosome conformation. Science 295 , 1306–1311 (2002).
Gheldof, N., Tabuchi, T. M. & Dekker, J. The active FMR1 promoter is associated with a large domain of altered chromatin conformation with embedded local histone modifications. Proc. Natl Acad. Sci. USA 103 , 12463–12468 (2006).
Service, R. Gene sequencing. The race for the $1000 genome. Science 311 , 1544–1546 (2006).
Cawley, S. et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell 116 , 499–509 (2004).
Tsang, J., Zhu, J. & van Oudenaarden, A. MicroRNA-mediated feedback and feedforward loops are recurrent network motifs in mammals. Mol. Cell 26 , 753–767 (2007).
Mangus, D., Evans, M. & Jacobson, A. Poly(A)-binding proteins: multifunctional scaffolds for the post-transcriptional control of gene expression. Genome Biol. 4 , 223 (2003).
Article PubMed PubMed Central Google Scholar
Kaern, M., Elston, T., Blake, W. & Collins, J. Stochasticity in gene expression: from theories to phenotypes. Nature Rev. Genet. 6 , 451–464 (2005).
Ule, J., Jensen, K., Mele, A. & Darnell, R. B. CLIP: a method for identifying protein–RNA interaction sites in living cells. Methods 37 , 376–386 (2005).
Golding, I., Paulsson, J., Zawilski, S. M. & Cox, E. C. Real-time kinetics of gene activity in individual bacteria. Cell 123 , 1025–1036 (2005).
Cai, L., Friedman, N. & Xie, X. S. Stochastic protein expression in individual cells at the single molecule level. Nature 440 , 358–362 (2006).
Raj, A., Peskin, C. S., Tranchina, D., Vargas, D. Y. & Tyagi, S. Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 4 , e309 (2006).
Bar-Even, A. et al. Noise in protein expression scales with natural protein abundance. Nature Genet. 38 , 636–643 (2006).
Newman, J. et al. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature 441 , 840–846 (2006). Reference 113 uses fluorescence-activated cell sorting (FACS) analysis of GFP–tagged proteins to examine cell-to-cell variability in the expression levels of different genes and to determine the extent to which certain mRNA characteristics contribute to noise in gene expression.
Blake, W. J., Mads, K. A., Cantor, C. R. & Collins, J. J. Noise in eukaryotic gene expression. Nature 422 , 633–637 (2003).
Becskei, A., Kaufmann, B. B. & van Oudenaarden, A. Contributions of low molecule number and chromosomal positioning to stochastic gene expression. Nature Genet. 37 , 937–944 (2005).
Raser, J. M. & O'Shea, E. K. Noise in gene expression: origins, consequences, and control. Science 309 , 2010–2013 (2005).
Bertone, P. et al. Global identification of human transcribed sequences with genome tiling arrays. Science 306 , 2242–2246 (2004).
Cheng, J. et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science 308 , 1149–1154 (2005).
Kampa, D. et al. Novel RNAs identified from an in-depth analysis of the transcriptome of human chromosomes 21 and 22. Genome Res. 14 , 331–342 (2004).
Willingham, A. T. & Gingeras, T. R. TUF love for 'junk' DNA. Cell 125 , 1215–1220 (2006).
Carninci, P. et al. The transcriptional landscape of the mammalian genome. Science 309 , 1559–1563 (2005).
Steinmetz, E. J. et al. Genome-wide distribution of yeast RNA polymerase II and its control by Sen1 helicase. Mol. Cell 24 , 735–746 (2006).
Struhl, K. Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nature Struct. Mol. Biol. 14 , 103–105 (2007).
Download references
Acknowledgements
The authors would like to thank D. Muzzey, M. Moore, I. Swinburne and C. Brown for helpful discussions and critical evaluation of the manuscript, and the support of grants from the US National Institutes of Health.
Author information
Authors and affiliations.
Department of Systems Biology, Harvard Medical School, Boston, 02119, Massachusetts, USA
Suzanne Komili & Pamela A. Silver
Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, 02119, Massachusetts, USA
Suzanne Komili
You can also search for this author in PubMed Google Scholar
Corresponding author
Correspondence to Pamela A. Silver .
Related links
Further information.
Pamela Silver's homepage
Histone variants share amino-acid sequence homology and core structural similarity with the canonical histone proteins but are functionally distinct. Some variants are restricted to certain regions of the chromosome, such as the centromere-specific H3-like variant CenpA. Others are used only during specialized processes, such as the histone H2A variant H2A.X, which binds to DNA with double-strand breaks and marks the region undergoing DNA repair.
The nuclear pore is a large protein complex that traverses the nuclear envelope, allowing transport between the nucleus and cytoplasm. The proteins that form the nuclear pore are known as nucleoporins.
The dosage compensation complex is an RNA–protein complex in Drosophila melanogaster that mediates the twofold hypertranscription of genes from the X chromosome in males, resulting in equalized levels of X-chromosome products in both sexes.
A synthetic genetic interaction occurs when the severity of the phenotype caused by the absence of two genes is more or less than the sum of the phenotypes of the individual gene knockouts. Two genes that are non-essential when deleted individually but for which the deletion of both is fatal are termed synthetic lethal.
Mutual information is a measure of dependence between two variables. If one variable is highly predictive of the other, then the mutual information between the two will be high. Importantly, this does not imply that the two variables exhibit the same behaviour, for anti-correlated variables are still co-dependent and thus have high mutual information.
Rights and permissions
Reprints and permissions
About this article
Cite this article.
Komili, S., Silver, P. Coupling and coordination in gene expression processes: a systems biology view. Nat Rev Genet 9 , 38–48 (2008). https://doi.org/10.1038/nrg2223
Download citation
Issue Date : January 2008
DOI : https://doi.org/10.1038/nrg2223
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
This article is cited by
Tobacco as green bioreactor for therapeutic protein production: latest breakthroughs and optimization strategies.
- Muhammad Naeem
- Lingxia Zhao
Plant Growth Regulation (2023)
Human transcription factor protein interaction networks
- Matias Kinnunen
- Markku Varjosalo
Nature Communications (2022)
Transcriptional competition shapes proteotoxic ER stress resolution
- Dae Kwan Ko
- Federica Brandizzi
Nature Plants (2022)
Transcriptionally induced enhancers in the macrophage immune response to Mycobacterium tuberculosis infection
- Elena Denisenko
- Sebastian Schmeier
BMC Genomics (2019)
Estimation of Transcription Factor Activity in Knockdown Studies
- Saskia Trescher
Scientific Reports (2019)
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing newsletter — what matters in science, free to your inbox daily.

- Reference Manager
- Simple TEXT file
People also looked at
Editorial article, editorial: coupling in biological systems: definitions, mechanisms, and implications.

- 1 Institute for Theoretical Biology, Humboldt University, Berlin, Germany
- 2 Computational Neuroscience Unit, Okinawa Institute of Science and Technology, Okinawa, Japan
- 3 Department of Mechanical Engineering, Ritsumeikan University, Shiga, Japan
- 4 Graduate Institute of Mind, Brain and Consciousness (GIMBC), Taipei Medical University, Taipei, Taiwan
- 5 Brain and Consciousness Research Centre (BCRC), TMU-Shuang Ho Hospital, New Taipei City, Taiwan
Editorial on the Research Topic Coupling in biological systems: Definitions, mechanisms, and implications
Biological systems exhibit an enormous complexity. Their temporal evolution ubiquitously depends on non-linear interactions between non-identical, heterogeneous entities such as molecules, cells, tissues, organs, or organisms. As a result, the observed dynamics at the ensemble level show emergent phenomena that can only occur at the macroscopic scale and cannot be understood solely from the intrinsic behavior of its individual constituents, limiting the success of traditional reductionist approaches ( Wolkenhauer and Green, 2013 ). The entities involved can exist in different positions of space and time and may span across various scales.
The process of interaction or information exchange between two or more entities in a given physical, biological or chemical system is often referred to as “coupling”. Using mathematical parlance, the dynamical evolution of a given part in a coupled system depends on the present or former state of other parts in the overall system.
In multicellular systems of neurons, the emergent dynamics is the neural network computation ( Figure 1 upper right ), governed by schemes of synaptic coupling (i.e., connectome). Likewise, the macroscopic output of physiology is a result of multi-organ coupling throughout the body ( Figure 1 middle ). At the organismic level, coupling between the environment and the body enables adaptation ( Figure 1 left ). On the lowest end of the biological hierarchy, coupling among molecular components creates feedback networks within a single cell ( Figure 1 bottom right ).

FIGURE 1 . Multiscale organization of circadian clock networks. Circadian clocks exist in all scales of biology, and so does the coupling among them. The scale spans from the whole organism (far left) to molecules within a single cell (bottom right).
In this Research Topic, we are particularly interested in coupled biological oscillators, i.e., systems composed of constituents that show rhythmically recurring patterns. Circadian oscillation is a ubiquitous biological phenomenon well suited for studying coupling across spatiotemporal scales. These self-sustained rhythms, with a period of approximately 1 day, are maintained in neuronal and non-neuronal systems at both cellular and organ levels.
An organism’s circadian rhythm must synchronize with the daily cycle most strongly defined by ambient light (called Zeitgeber, or “time-giver”). This uni-directional coupling between a Zeitgeber and a forced pacemaker is the simplest form of coupling, yet it can yield an astonishing variety of dynamical phenomena ( Heltberg et al., 2021 ), in particular in response to seasonal variations of day length and luminosity ( Schmal et al., 2020 ; Burt et al., 2021 ). Healy et al. extend the concept of Zeitgeber to include non-photic cues such as sleep, feeding-fasting cycles, or physical activity. A host of molecular pathways are suggested, which can independently and/or synergistically entrain the clock through different molecular components such as PER1/2, CRY1/2, BMAL1/CLOCK, and DBP. Grabe et al. mathematically explore this in a two-oscillator system simultaneously entrained by photic and non-photic Zeitgebers. The molecular circadian clock network is composed of two main loops of transcriptional-translational feedback: One centers on transcriptional regulation of E-box (controls, notably, Per/Cry ) and the other on RRE (drives transcription of Bmal1 ) elements. Mammals have a central circadian clock that resides in the hypothalamic suprachiasmatic nucleus (SCN), which receives light signals directly from the retina. Using an evolutionary game theoretic framework, Spencer et al. seek for coupling topologies of the SCN that sustain circadian synchronization at minimal “cost”. The evolutionary mechanism can drive the SCN network to adopt sparse coupling against the metabolically expensive all-to-all coupling. Gu et al. consider the heterogeneous structure of the SCN network that can be divided into a dorsomedial (DM; shell) and a ventrolateral (VL; core) subregion. The authors suggest the possibility that the sparse network of the SCN can attain synchronization through small-world/scale-free coupling. The brain contains several circadian clock loci outside the SCN. Chrobok et al. discuss coupling in one stream of these clocks along the subcortical visual system (SVS). The SVS exhibits various timescales of neural firing, and circadian coupling is thought to enable multiplexing different frequency bands.
The circadian system provides an ideal context to study coupling in biological systems, which maintain oscillations in various scales of space and time. Although coupling is often a conceptualization of indirect interactions that simplifies a series of underlying processes, its consequence can be directly explored through mathematical modeling to produce experimentally testable predictions. Mathematical modeling can also provide a platform to classify the strength, directionality, and polarity of coupling in a wide range of spatiotemporal dynamics of biology while being neutral to exact details.
Author contributions
CS and JM drafted the manuscript. CS created the figure and JM revised it. All authors reviewed and agreed on the manuscript.
CS acknowledges support from the Deutsche Forschungsgemeinschaft (DFG) through grant SCHM3362/2-1—Project-ID 414704559. JM is financially supported by the Higher Education Sprout Project by the Ministry of Education (MOE) and in part by National Science and Technology Council (NSTC) in Taiwan under grants 110-2311-B-038-003, 110-2314-B-038-162, and 111-2314-B-038-008.
Acknowledgments
We thank Anna-Marie Finger for the initial arrangements of the Research Topic.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Burt, P., Grabe, S., Madeti, C., Upadhyay, A., Merrow, M., Roenneberg, T., et al. (2021). Principles underlying the complex dynamics of temperature entrainment by a circadian clock. iScience 24 (11), 103370. doi:10.1016/j.isci.2021.103370
PubMed Abstract | CrossRef Full Text | Google Scholar
Heltberg, M. L., Krishna, S., Kadanoff, L. P., and Jensen, M. H. (2021). A tale of two rhythms: locked clocks and chaos in biology. Cell Syst. 12 (4), 291–303. doi:10.1016/j.cels.2021.03.003
Schmal, C., Herzel, H., and Myung, J. (2020). Clocks in the wild: entrainment to natural light. Front. Physiol. 11, 272. doi:10.3389/fphys.2020.00272
Wolkenhauer, O., and Green, S. (2013). The search for organizing principles as a cure against reductionism in systems medicine. FEBS J. 280, 5938–5948. doi:10.1111/febs.12311
Keywords: circadian clock, coupling, biological network, entrainment, zeitgeber, suprachiasmatic nucleus (SCN)
Citation: Schmal C, Hong S, Tokuda IT and Myung J (2022) Editorial: Coupling in biological systems: Definitions, mechanisms, and implications. Front. Netw. Physiol. 2:1076702. doi: 10.3389/fnetp.2022.1076702
Received: 21 October 2022; Accepted: 30 November 2022; Published: 13 December 2022.
Edited and reviewed by:
Copyright © 2022 Schmal, Hong, Tokuda and Myung. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christoph Schmal, [email protected] ; Jihwan Myung, [email protected]
This article is part of the Research Topic
Coupling in Biological Systems: Definitions, Mechanisms, and Implications
- BiologyDiscussion.com
- Follow Us On:
- Google Plus
- Publish Now
Coupling and Repulsion Hypothesis of Gene

ADVERTISEMENTS:
The below mentioned article provides a short note on the Coupling and Repulsion Hypothesis of Gene.
Bateson and Punnett in 1906, described a cross in sweat pea, where failure of gene pairs to assort independently was exhibited. Plants of a sweat pea variety having blue flower (BB) and long pollen (LL) were crossed with those of another variety having red flower (bb) and round pollen (II). F 1 individuals (BbLl) had blue flower and long pollen.
These were test crossed with plants having red flower and round pollen (bbll).
In this case, the character for blue colour of flower is dominant over red colour, and long pollen character is dominant over round pollen. In case of independent assortment, one should expect 1:1:1:1 ratio in test cross. Instead 7:1:1:7 ratio was actually obtained, indicating that there was a tendency of dominant alleles to remain together.
The case was similar for recessive alleles also. This deviation was explained in terms of gametic coupling by Bateson.
It was also observed that when two such dominant alleles or recessive alleles come from different parents, they tend to remain separate. This was termed as gametic repulsion. In Bateson’s experiment, in repulsion phase, one parent would have blue flower and round pollen (BBII) and the other would have red flower and long pollen (bbLL).
The results of test cross in such a repulsion phase were similar to those obtained in coupling phase, giving 1:7:7:1 ratio instead of expected 1:1:1:1 (Fig. 8.1).

Bateson explained the lack of independent assortment by means of a hypothesis known as coupling and repulsion hypothesis. Later on, coupling and repulsion were discovered to be two different aspects of linkage.
Related Articles:
- Biology Notes on Mendelian Inheritance | Genetics
- Linkage of Genetics: Features, Examples, Types and Significance
Notes , Biology , Cell Biology , Gene Mapping , Gene , Coupling and Repulsion Hypothesis
- Anybody can ask a question
- Anybody can answer
- The best answers are voted up and rise to the top
Forum Categories
- Animal Kingdom
- Biodiversity
- Biological Classification
- Biology An Introduction 11
- Biology An Introduction
- Biology in Human Welfare 175
- Biomolecules
- Biotechnology 43
- Body Fluids and Circulation
- Breathing and Exchange of Gases
- Cell- Structure and Function
- Chemical Coordination
- Digestion and Absorption
- Diversity in the Living World 125
- Environmental Issues
- Excretory System
- Flowering Plants
- Food Production
- Genetics and Evolution 110
- Human Health and Diseases
- Human Physiology 242
- Human Reproduction
- Immune System
- Living World
- Locomotion and Movement
- Microbes in Human Welfare
- Mineral Nutrition
- Molecualr Basis of Inheritance
- Neural Coordination
- Organisms and Population
- Photosynthesis
- Plant Growth and Development
- Plant Kingdom
- Plant Physiology 261
- Principles and Processes
- Principles of Inheritance and Variation
- Reproduction 245
- Reproduction in Animals
- Reproduction in Flowering Plants
- Reproduction in Organisms
- Reproductive Health
- Respiration
- Structural Organisation in Animals
- Transport in Plants
- Trending 14
Privacy Overview

- school Campus Bookshelves
- menu_book Bookshelves
- perm_media Learning Objects
- login Login
- how_to_reg Request Instructor Account
- hub Instructor Commons
- Download Page (PDF)
- Download Full Book (PDF)
- Periodic Table
- Physics Constants
- Scientific Calculator
- Reference & Cite
- Tools expand_more
- Readability
selected template will load here
This action is not available.
1.5: Linkage
- Last updated
- Save as PDF
- Page ID 106368
- Thomas Lübberstedt, Laura Merrick, Deborah Muenchrath, Arden Campbell, & Shui-Zhang Fei
Introduction
Genes located on the same chromosome are genetically linked. Genetic linkage analysis can be used to determine the order of genes on chromosomes. Closely linked genes are not segregating independently, like genes located on different chromosomes. This has different implications, e.g., in relation to trait correlations. Moreover, linked genes can be used as genetic markers, which have become an important tool in plant breeding.

Learning Objectives
- Develop an understanding of the genetic basis of linkage.
- Gain awareness on how to detect the occurrence of linkage.
- Review the principles of genetic map construction.
- Become familiar with the concept of linkage disequilibrium.
Crossover and Recombination
Genetic organization.

Genes are physically organized on chromosomes. Each gene is located at a particular “address” (particular position on a specific chromosome, which can be identified by genetic mapping). Inheritance of genes located on different chromosomes follows the rules of independent assortment. Since plant species have multiple chromosomes, independent assortment is true for the majority of genes. In contrast, linked genes located on the same chromosome are more likely to cosegregate, i.e., being jointly transmitted to offspring more often than expected by independent assortment. The biological process that separates linked genes is the crossing-over (C.O., or crossover), which occurs during meiosis, and leads to genetic recombination.
Crossing-Over
Query \(\pageindex{1}\).
During meiosis of diploid organisms, the chromatids of homologous chromosomes pair and form bivalents. During Meiosis I, homologous chromatids pair to physically exchange chromosome segments. The chromosomal site, where this reciprocal exchange of homologous chromosome segments takes place, is called a chiasma . Thus, crossing-over involves not completely understood mechanisms for identification of homologous sites of chromatids, breakage and rejoining of chromosomes.
Genetic Distance
Crossing-over events occur more or less random during meiosis. In most plant species, one to few crossing-over events occur per meiosis and chromosome. Thus, the closer the genes are physically linked on the same chromosome, the less likely they will get separated, and consequently, the less likely genetically recombinant gametes will be produced. This is the underlying principle of genetic maps: the genetic distance between genes reflects the probability of a crossing-over between linked genes.
Query \(\PageIndex{2}\)

Recombination
Observation of crossing-over events requires cytological methods, which can be cumbersome for large populations. In contrast, genetic recombinants can be observed at the phenotype level, or by use of DNA markers. If two linked genes with two alleles each have clear phenotypic effects, e.g., on flower color ( A : red, a : white; A is dominant over a ) and seed color ( B : green, b : yellow; B is dominant over b ), then genetic recombinants can easily be identified by determining the fraction of non-parental gametes in the offspring.
Note that crossing-over also takes place in meiosis of completely homozygous individuals. However, in this case, genetic recombination cannot be observed as described above. The reason is that observation of recombinant gametes requires two (or more) different alleles at the loci, for which linkage is going to be determined. This explains why offspring saved from pure line cultivars will not segregate whereas seed harvested from F 1 hybrid will segregate. The observable fraction of recombination events is also called effective recombination .
Linkage Detection
Linkage phase.
For linkage detection, it is crucial to know the linkage phase of alleles.
The linkage phase is the physical arrangement of linked genes in a chromosome. A double heterozygote with a genotype of AaBb could be in one of the two linkage phases. Conventionally, when linked dominant alleles are located on the same homologous chromosome and the linked recessive alleles are on the other homologous chromosome, for example, AB/ab , it is said the genes are linked in the coupling phase. When a dominant allele at one locus is on the same homologous chromosome as a recessive allele of the other linked gene, for example, Ab/aB , it is said that the genes are linked in repulsion phase (Figure 4).
This knowledge is crucial, as linkage detection and distance estimation is based on the observed parental and non-parental gametes.

Coupling and Repulsion
In the case of close linkage, non-parental gametes and respective offspring are underrepresented.
An example is the Australian sheep blowfly, Lucilia cuprina . Normal blowflies have a green thorax and surround themselves in a brown cocoon during their pupal stage. However, recessive genes (here marked a and b ) can cause the fly to develop a purple thorax and spin a black puparium.

Query \(\PageIndex{3}\)
Using testcrosses.
For detection of linkage, appropriate testcrosses need to be conducted. The linkage phase is known, if two homozygous parental genotypes ( AABB and aabb ) are crossed to produce the respective F 1 ( AaBb ).

In this case, A and B as well as a and b are linked in coupling phase.
The non-parental recombinant gametes have the genotype Ab and aB , whereas the parental gametes have the genotype AB and ab .
Usually the phenotype cannot be observed in (haploid) gametes, but only in diploid plants. Thus, to determine whether two loci are linked, offspring need to be produced. This can be achieved by self pollination of the AaBb – F1, by production of doubled haploid offspring, or by a testcross.
In this particular example, a backcross (BC) of the F 1 to the aabb parent would be the best option.
Testcross Gametes

All offspring from this BC would receive an ab gamete from the aabb parent, and any of the two parental ( AB , ab ) or non-parental ( Ab , aB ) gametes from the F 1 .
Because of dominance of A over a and B over b , all four resulting diploid genotypes in the BC 1 (backcross generation 1) generation ( AaBb , Aabb , aaBb , aabb ) can be phenotypically discriminated, and used to count genotypes that received parental or non-parental gametes from the F 1 .
Thus, when using this BC approach, only the crossing-over events that occurred in the F 1 are monitored for linkage estimation.
Chi-Square Test
For detection of linkage, a Chi-Square test can be employed. The Chi-Square test compares observed with expected frequencies. In this case, the null hypothesis to determine expected frequencies is the assumption of independent assortment. Under this assumption, equal frequencies of all four gametes are expected. In the case of linkage, BC1 individuals carrying non-parental gametes are underrepresented, leading to a statistically significant Chi-Square value. This means that the null hypothesis of independent assortment would be rejected and linkage assumed.
Chi-Square Results
To better understand the use of Chi-Square in determining linkage, two numerical examples based on the cross schemes described in Figs. 8 and 9 are provided here. In both examples, a sample size of 2,000 BC1 individuals has been used.
The Chi-Square test sums up over all squared differences between observed and expected values, divided by expected values.
In example A, observed and expected values are equal, thus the Chi-Square value = 0.

In example B, the squared differences between observed and expected values is in all cases 90,000, to be divided by the expected 500 = 180. As there are four genotypic classes, the Chi-Square value is 720, which is significantly larger than the tabulated value of 3.81 (p = 5%).
In conclusion, example A is in agreement with independent assortment, whereas in example B, linkage has been detected.

The same data used to determine linkage can also be used to estimate the recombination frequency between two genes (more precisely, the recombinant frequency). The recombinant frequency = (number of BC1 progeny with recombinant (non-parental) alleles / total number of BC1 progeny) x 100%.
In example B of Section 2: Linkage Detection, the recombinant frequency is (200 + 200 / 2000) * 100% = 20%.
The recombination frequencies between any pairs of genes provide an estimate of how close they are linked on a chromosome. The recombinant frequency in % is sometimes also called “map units” (M.U.). In this example, the genetic distance in map units between the two genes under consideration is 20 M.U.
In the case of complete linkage of two genes, no recombinants would be expected. The recombinant frequency would be 0%, which represents the lower limit of recombinant frequencies.
In the case of random segregation, the expected numbers of recombinant and nonrecombinant alleles are equal. Thus, the upper limit of recombinant frequencies in the case of unlinked or loosely linked genes is 50%.
Even for gene pairs located at the different ends of the same chromosome, recombination frequency can reach 50%. The procedure to determine recombination frequencies between any pair of genes is called two-point analysis.
You have a F 1 plant heterozygous at two loci that are 12 map units apart on the same chromosome. The F 1 received linked recessive alleles from one parent and linked dominant alleles from the other parent.
Query \(\PageIndex{4}\)
Since linkage cannot be detected in the F 1 , you self-pollinate the F 1 and evaluate the F 2 . What would be the F 2 and testcross percentages if the F 1 percentages in the case of repulsion phase of the recessive alleles?
Query \(\PageIndex{5}\)
Three-point analysis.
Whereas two-point testcrosses establish linkage between pairs of genes, three-point testcrosses facilitate establishment of the order of genes on chromosomes, as a prerequisite to establishing genetic maps. If a third locus with alleles C and c ( C is dominant over c ) is added to the case mentioned in Genetic Distance, where A and B are linked in coupling phase and the dominant allele C is in coupling with A and B , then eight different testcross progeny would result from a backcross with the recessive parent.

Frequency Chart
Pairwise recombination frequencies can be determined as described in the chart.
- 20.8% for AC ( AC recombinants are in classes 3, 4, 7, and 8; thus, the recombination rate between A and C is (52+46+4+2/500) * 100% = 20.8%)
- 10% for CB ( CB recombinants are in classes 5-8)
- 28.4% for AB ( AB recombinants are in classes 3-6).
Once linkage between pairs of three (or more) genes has been established, the next question is how they are arranged in linear order on chromosomes, which could be ABC , ACB , or CAB .
The most likely gene order minimizes the sum of pairwise recombination frequencies within a three-gene interval, which would be:
- 38.4 for ABC (28.4% for AB + 10% for CB )
- 30.8 for ACB (20.8% for AC + 10% for CB )
- 49.2 for CAB (20.8% for AC + 28.4% for AB )
Thus, the most likely gene order is ACB . In other words, the interval between A and B can be subdivided into the intervals between AC and CB .
Expressed yet another way: incorrectly ordered genes would increase the total map length because part of the recombination events would be counted twice. If ACB is the true order, then the genetic length of, e.g., ABC would be inflated, because recombinants for the segment BC would be counted two times: for the interval BC , in addition to the same interval within the segment A(C)B . Algorithms of mapping programs use this principle (minimizing the genetic distance) for three-point analyses.
Double Crossovers
The two-point recombination frequency between A and B (28.4%) differs from the sum of recombination frequencies for AC and CB (30.8%). The reason for this discrepancy is the occurrence of double crossover events. These are two crossovers in a single meiosis within an interval of interest and the second crossover reverses the effect of the first crossover, i.e., the second crossover returns the B allele to the original position before the first crossover. For this reason, by only taking recombinants between A and B into consideration, double crossovers cannot be observed. Because a double crossover exchanges chromosome segments within an interval of two genes, the linkage phase (coupling) of those two genes remains unchanged.
Observing Double Crossovers
Only by adding a gene like C in between A and B , it is possible to observe double crossovers. In the case of the interval between genes A and B , six double crossovers were observed. In consequence, recombination and crossover frequencies are not identical. The larger the genetic interval, the larger the discrepancy between recombination and crossover frequencies, because even-numbered crossover events within a pair of genes go undetected. By adding an additional gene in this interval, at least some double crossovers can be detected. This leads to detection of additional recombination events. For this reason, the recombination frequency between A and B in the example is increased, after adding C in between those two genes, because 6 double crossovers (= 12 additional recombination events) could be detected. Those 12 additional detectable recombination events explain for the 2.4% difference between recombination frequencies detected for the gene pair A and B with or without inclusion of C .
Phase Analysis
Recombinants resulting from double crossovers are always in the lowest frequency (class 7 and 8, respectively in this table). To determine which allele is in the middle, a convenient method is to find out which allele in the double crossover recombinants has changed its linkage phase with the other parental alleles (in classes 7 and 8, allele C/c has changed its linkage phase with the other alleles).
Coefficient of Coincidence and Interference
Crossover events in adjacent chromosome regions might affect each other, a phenomenon called interference . Most typically, a crossover event in one region tends to suppress a crossover in the adjacent regions. The extent of interference is expressed by the coefficient of coincidence , which is equal to the observed frequency of double crossovers / expected frequency of double crossovers.
The expected frequency of double crossovers is the product of two single crossovers in adjacent regions assuming there is no interference.
In this example, this expected frequency is 0.21 (recombination frequency for AC) * 0.10 (recombination frequency for CB) = 0.021.
The observed frequency of double crossover events is 6/500 in the example, resulting in 0.012. Thus, the coefficient of coincidence in this example is 0.012/0.021 = 0.58.
Interference is defined as 1 − coefficient of coincidence, which would be 0.42 in this example. A value of zero for interference would mean that a crossover in one region does not affect crossovers in the adjacent region. Interference of 1 means, that crossovers in one region suppress crossovers in the adjacent region. Negative values are possible and have been reported in some instances, which means that crossovers in one region stimulate crossovers in the adjacent region.
Map Functions
Measurement units.
The purpose of genetic maps is to report the length of chromosome intervals, chromosomes, and whole genomes. Since recombination frequencies converge to a value of 50% as reported above, indicating the absence of linkage, recombination frequencies are not additive and, thus, not useful to describe the distance between genes that are located far apart. When recombination frequency reaches 50%, it would be impossible to tell whether the genes are located far apart on the same chromosome or on different chromosomes.
Instead, estimates of the number of crossover events are used as an additive measure of genetic map distances. The unit for measuring genetic distances is Morgan (M), or usually centiMorgan (cM) . In contrast to recombination frequencies, map units expressed in cM are additive. One Morgan reflects the observation of one crossover event per single meiosis. One cM is a distance between genes that produces 1% recombinants in the offspring. Typical lengths of genetic maps in maize, for example, vary between 1,600 to 2,000 cM, which means that on average, 1.6 – 2 crossovers occur per chromosome and single meiosis in maize (maize has 10 homologous chromosome pairs).
Frequency Conversion
As mentioned above, direct observation of crossover events is cumbersome. For that reason, most genetic maps published to date are based on the conversion of recombination frequencies into crossover frequencies. The main obstacle to translating recombination frequencies into crossover frequencies is the variable and unknown degree of interference in different genome regions. While it has been possible in the earlier example to determine the degree of interference, and thus frequency, of double crossover events, in the genetic interval between A and B by adding C , the degree of interference between AC and CB is unknown. This could be addressed by observing segregation of further genes within these two regions (if available), but this issue could ultimately only be addressed by complete genome sequencing of all offspring in a mapping population, which at this point is still too costly.
Visual Relationship
Instead, map functions have been developed, that translate recombination frequencies into crossover frequencies, and thus cM (see Figure 14 below). Figure 14 clearly shows, that there is an approximately linear relationship between recombination rates (y-axis) and crossover rates (x-axis). However, with increasing map distances, recombination rates converge to 50%. In other words, gene pairs with crossover rates of 80 cM or 200 cM, respectively, would be nearly indistinguishable based on recombination rates, which would result in recombination rates between 40 and 50%.
The various available map functions make different assumptions on the extent of interference. For example, the Haldane mapping function assumes absence of interference. In contrast, the Kosambi function assumes the presence of interference.
Other Types of Maps
Genetic maps can be generated in other ways than using testcrosses. Examples include somatic cell hybridization and tetrad analysis. In plants, interspecies addition lines such as oat-maize addition lines created by distant hybridization have been developed as tool for mapping of genes. If two genes appear on the same addition segment, they are genetically linked. Besides genetic maps, cytological and physical maps can be established.

Cytological maps show gene orders along each chromosome as determined by cytological methods whereas physical maps are measured in base pairs as determined by DNA sequencing. With rapid progress in sequencing technology and an increasing number of sequenced plant genomes, physical maps gain in importance. Complex plant genomes like the maize genome are billions of base pairs long. When comparing genetic and physical maps, the order of genes is conserved. However, the relative distances between genetic and physical maps might vary substantially. The reason is that crossover events are not evenly distributed in genomes. Usually, crossover events tend to be suppressed in centromere and repetitive DNA regions, whereas they are enhanced in gene-rich regions.
Cytogenic Map

Factors Influencing Linkage Mapping
Linkage mapping based on testcrosses can be affected by selection or incomplete penetrance, among others. Selection in the most extreme case would be due to lethality of gametes (gametic selection) or zygotes (zygotic selection). If a backcross is used for linkage detection, as described above, lethality of male gametes carrying for example the a allele would lead to only two classes of BC progeny, if AaBb is crossed as pollinator to aabb . In that case, only AaBb (parental) and Aabb (recombinant) genotypes would be obtained. Zygotic selection affects the viability of particular genotypes. If the aa genotype in the example above is lethal, then the aa offspring derived from self-pollination of an AaBb genotype would be missing. Incomplete penetrance means that a genotype that is supposed to express, for example, red flowers, has to a certain extent white flowers. In other words, there is no 100% match between genotype and phenotype, but due to environmental factors, the phenotype might differ. As for selection, incomplete penetrance alters the frequency of expected genotypes in testcrosses, which is the basis for detection of linkage.
Consequences and Applications of Linkage
The main application of linkage is in genetic mapping of genes using molecular markers. Once genes have been mapped and closely-linked markers identified, those markers can be used for marker-aided selection procedures. Technological progress in DNA methods has been and still is rapid, so that thousands of markers can be produced at low cost in any species of interest. Moreover, novel genomic selection strategies addressing complex inherited traits are being developed.
Linkage can in some cases be confused with pleiotropy. If a favorable character (e.g. resistance) is always inherited together with an unfavorable trait (e.g., lodging), a negative pleiotropic effect might be assumed, which might alternatively be caused by two closely linked genes. Whereas close linkage can be resolved to find favorable genotypes for both traits, this is not true for pleiotropy. Linkage reduces the possible genetic variation in small populations. With increasing numbers of generations, or population sizes, genetic variation can be increased. Similarly, inbreeding reduces the opportunity for effective recombination.
Linkage Disequilibrium
Genotype distribution.
Although allele frequencies at individual loci are expected to be stable in the case of random mating, genotype frequencies at two or more loci jointly do not achieve this equilibrium after one generation of random mating.
}{2})^k")
Equilibrium
Consider the following examples based on two alleles at each of two loci:
- Allele frequencies : P A P a P B P b
- Gametic Frequencies : P AB P Ab P aB P ab
In linkage equilibrium, the expected gamete frequencies can be calculated from the marginal allele frequencies. For example, in equilibrium, the frequency of gamete AB (P AB ) would be expected to be equal to the product of the frequencies of the A allele (P A ) and the B allele (P B ).
This is valid under the following conditions: P A + P a = 1; P B + P b = 1; and P AB + P Ab + P aB + P ab = 1.
If, for example, the allele frequencies of P A = P a and P B = P b are 0.5, then the frequencies of all gametes are 0.25.
A measure for Disequilibrium, D = P AB – P A *P B . D = 0 in the case of equilibrium. If D differs from 0, it reflects presence of Disequilibrium. In other words, the frequency of a gamete differs from its expected frequency based on marginal probabilities of the respective individual alleles.
LD and Mapping
Linkage disequilibrium is the non-random association of alleles at different loci. LD is extensively used in mapping human disease genes using natural populations (Association mapping).
In plants, gene mapping has been conducted mainly by using mapping families because of the ease with which mapping families are created, but LD mapping using natural populations is increasing rapidly because such populations are large in size and have much greater allelic diversity.

LD Statistic D’
}")
for DAB < 0
}")
for DAB > 0
Dissipation
It can be shown that after t generations of random mating, the remaining disequilibrium is given by:
\[D_t = D_0 (1 - c)^t\]
where, D 0 is the disequilibrium in generation 0 and c is the recombination fraction, with c = 0.5 for independently segregating loci, which is identical to a recombination frequency of 50% (the range of c is from 0 to 0.5, whereas the range of r is from 0% to 50%). The dissipation of disequilibrium relative to generation 0 is given in Figure 18.
Query \(\PageIndex{6}\)
Recombination and ld.
Generally, deviations from independence at multiple loci are referred to as linkage disequilibrium, even if genetic linkage is not the cause (in other words, alleles are not physically linked). Unless two loci are known to reside on the same chromosome, the term Gametic Disequilibrium should be used to describe disequilibrium among loci. Whereas recombination and crossover frequencies, as mentioned initially, are used to describe the distance between genes from a chromosomal perspective, linkage disequilibrium is mostly used to describe a property of populations. However, both terms are closely related.
Genetic Markers
Genetic variation results from differences in DNA sequences and, within a population, occurs when there is more than one allele present at a given locus. Such populations are referred to as populations that are polymorphic or segregating at that locus. The opposite situation is when all members of the population are homozygous for the same allele, in which case the population is said to be fixed or monomorphic for that allele. A genetic marker is a DNA sequence that exhibits polymorphism among individuals and can thus be used to identify a particular locus (although not necessarily a gene) on a particular chromosome; the marker itself may be part of a gene or may have no known function. Markers are inherited in a Mendelian fashion and facilitate the study of inheritance of a trait or sometimes a linked gene. Markers are used to identify, map , and isolate genes, select desired genotypes, and detect genetic variation or determine genetic relationships among individuals. Markers are regions of genomes that are heritable, often easy to document, and useful for detecting genetic variation.
Three Types
Genetic markers generally do not represent target genes of interest to a breeding program, but instead are useful as ‘signs’ or ‘tags’, particularly when they are closely linked to genes that control a trait of interest. A genetic map constructed with genetic markers is similar to a road map. Linkage groups in a genetic map represent roads whereas individual markers on each linkage group represent signs or landmarks that help plant breeders to navigate through the plant genome and find the genes of interest.
There are three major categories of markers.
- Morphological markers
- Biochemical markers
- Molecular markers
Morphological Markers
These types of markers (also called visible or classical markers) are phenotypic traits with only a few distinct morphs or variants (e.g., flower color or seed shape), usually due to one or perhaps two gene loci so they are not strongly affected by the environment. Inheritance patterns of visible and morphological characters have been used to map genes to particular chromosome segments and to identify linkage groups. Such markers are limited in number compared to the abundance of DNA markers, however, and may be influenced by developmental stage of the plant.
Biological Markers
Isozymes (sometimes called allozymes) are allelic variants of a single enzyme that share the same function, but may differ in level of activity due to differences in amino acid sequence. Isozymes are proteins for which variation can be detected by differential separation using electrophoresis , a technique for separating macromolecules (DNA, RNA, protein) on a gel by means of an electric field and specific chemical staining.
Isozymes have codominant expression, meaning that both homozygotes can be distinguished from the heterozygote and neither allele is recessive. In contrast to codominant markers, dominant markers are either present or absent.
In comparison to visible polymorphisms, they reveal more of the underlying genetic variation. However isozymes are gene products, so they reveal only a small subset of the actual variation in DNA sequences between individuals and do not reveal variation in the non-coding regions of the genome. In general, such markers are limited in number and have limited use in genetic mapping studies.

Molecular Markers
Molecular or DNA markers reveal sites of variation in DNA. Variability in DNA facilitates finer scale mapping and detection. Mapping is the process of making a representative diagram cataloging genes and other features of a chromosome and showing their relative positions. Many of these molecular markers avoid the limitations associated with visible and biochemical markers. They facilitate the evaluation of genome-wide coverage and are not affected by environmental factors or developmental stages. They allow high resolution of genetic diversity to be detected. Molecular markers have added substantial amounts of information to our genetic maps.
FYI: Molecular Markers
Any DNA sequence can be genetically mapped, like genes leading to plant phenotypes as long as there is a polymorphism available for the sequence to be mapped, i.e., two or more different alleles. This can basically be a single nucleotide polymorphism (SNP) , a single nucleotide variant at a particular position within the target sequence, or an insertion/deletion ( INDEL ) polymorphism. Any target sequence can be amplified by the Polymerase chain reaction (PCR) , and subsequently be visualized to generate “molecular phenotypes” comparable to visual phenotypes, that can be observed by using appropriate equipment.

Various molecular methods have been developed to visualize SNPs or INDEL polymorphisms at low cost and high throughput, which will be presented in detail in the Molecular Genetics and Biotechnology course. The main use of those SNPs and INDEL polymorphisms is as molecular markers. By genetic mapping as described above, linkage between genes affecting agronomic traits or morphological characters, and DNA-based SNP or INDEL markers can be established. It can be more effective in the context of plant breeding, to select indirectly for such DNA markers, than directly for target genes. This is due to lower costs for DNA analyses, the ability to run multiple such assays (for multiple target genes) in parallel, the ability to select early and to discard undesirable genotypes or to perform selection before flowering, codominant inheritance of markers, among others.For example, both ginkgo trees ( Ginkgo biloba ) and asparagus ( Asparagus officinalis ) are dioecious species. Male plants are preferred for ginkgo tree because fruits produced from female trees have an unpleasant smell whereas male asparagus plants are preferred because of their higher yield potential. Unfortunately, sex expression will take years to occur for both species. If a DNA marker that either directly affects sex expression or is linked to genes that affect sex expression can be identified, selection of male plants can be conducted in early seedling stage rather than waiting for many years. Occurrence of environmental conditions favoring selection for disease, insect-resistant plants or drought-tolerant plants such as the prevalence of the particular disease or insect or drought is not always reliable. Selection using DNA markers can overcome these limitations as they are not affected by the environment.
Polymorphism
Polymorphism involves one of two or more variants of a particular DNA sequence. The most common type of polymorphism involves variation at a single base pair, also called single nucleotide polymorphism (SNP) (Figure 22). Polymorphisms can also be much larger in size and involve long stretches of DNA. Tandem repeat is a sequence of two or more DNA base pairs that is repeated in such a way that the repeats are generally associated with non-coding DNA. In contrast, SNPs can sometimes be identified that occur within coding sequences (that is within genes), as well as in non-coding DNA.

Types of Biochemical/Molecular Markers
There are a variety of biochemical and molecular markers available. Table 2 on the next page summarizes features of a number of the common ones:
- RFLP — Restriction Fragment Length Polymorphisms
- RAPD — Random Amplified Polymorphic DNA
- AFLP — Amplified Fragment Length Polymorphisms
- SSR — Simple Sequence Repeats (also known as microsatellites )
- SNP — Single Nucleotide Polymorphisms
- VNTR — Variable Number of Tandem Repeats
Widely-Used Markers
* ‘PCR’ means Polymerase Chain Reaction amplification of genomic DNA fragments, a method that uses short, single-stranded DNA sequences, known as primers, to hybridize with the sample DNA Table 2. Comparison among widely used molecular markers. Adapted from Nageswara-Rao and Soneji, 2008.
SSR and SNP Markers
SSR markers remain useful to plant breeders due to their abundance and convenience with which they are assessed, but they serve most likely as linked markers. SNP markers, however can either be linked to or directly reside in a gene of interest and are hugely abundant. For these reasons, they are increasingly becoming the marker of choice.
Uses of Molecular Markers
Molecular markers are useful for both applied and basic genetic research. Here are some examples:
Indirect selection criteria in breeding programs (marker-assisted selection)
This is one of the most important and widely used molecular techniques in applied plant breeding programs today. RFLPs, SSRs, and SNPs enable breeders to indirectly select for a desired trait. Ordinarily, the DNA sequence of a molecular marker does not itself code the gene for the trait, but rather, its presence is correlated or linked with the gene for the particular trait. Thus, the breeder can indirectly select for the trait by directly selecting for the molecular marker—the DNA fragment and the gene encoding the trait are linked. The closer their physical proximity on a chromosome, the greater the probability that they will remain linked and not be separated through a recombination event in subsequent generations. As long as the gene for the trait and the marker remain linked, the marker is a useful selection criterion. Ultimately, however, potential lines still must be field-tested to verify the expression of the desired phenotype.
Identify quantitative trait loci (QTLs)
Molecular markers linked to genes contributing to the expression of polygenic or quantitative traits can be used to more efficiently identify and select individuals possessing the genes. It is more difficult to use conventional breeding approaches to identify plants that have accumulated the genes necessary to obtain the desired quantitative trait.
Genetic mapping
Molecular markers provide a means to map genes to more specific chromosome segments than is possible using visible markers.
Determine genetic relationships
The more molecular markers individuals have in common, the more closely related they are.
Genetic Diversity and Conservation
Genetic relationships within families, genera, species, or cultivars can be determined from molecular markers. Much information about the evolution of crops has been learned using molecular markers. The markers also enable breeders to monitor the genetic diversity among breeding lines to broaden the genetic base and reduce the risk of widespread genetic vulnerability to detrimental conditions.
Molecular Marker ‘Fingerprints’
Individuals possessing more markers in common than could occur by random chance are closely related. Such molecular fingerprints have been used successfully in court to prove the misappropriation of proprietary breeding lines.
Isolate genes
Molecular markers are used to map candidate genes on a much finer scale and can eventually isolate candidate genes by positional cloning. Isolated genes can be used to study gene regulation or to directly improve agronomic performance by genetic transformation. Although molecular markers have many applications and provide useful tools to plant breeders, lines must still be evaluated under normal production conditions before their release.
Falconer, D.S., and Trudy F.C. Mackay. Introduction to Quantitative Genetics (4th edition). San Francisco, CA: Benjamin Cummings.
Fehr, Walter R. 1987: Principles of Cultivar Development. Macmillan Publishing Company: New York.
Nageswara-Rao, M., J.R. Soneji, C. Chen, S. Huang, and F.G. Gmitter. 2008. Characterization of zygotic and nucellar seedlings from sour orange-like citrus rootstock candidates using RAPD and EST-SSR markers. Tree Genet Genomes. 4:113-124. DOI: 10.1007/s11295-007-0092-2
Neuffer M.G., Coe, E.H. and Wessler, S.R. (1997) Mutants of Maize. Plainview, NY: Cold Spring Harbor Laboratory Press
NIH-NHGRI (National Institutes of Health. National Human Genome Research Institute). 2011. Talking Glossary of Genetic Terms. [available online September 23, 2011, http://www.genome.gov/glossary/ .
Pierce, Benjamin A. 2008: Genetics: A Conceptual Approach. W.H. Freeman and Company: 160-199, 335-339. New York, NY.
Rafalski, A. 2002 Applications of single nucleotide polymorphisms in crop genetics. Current Opinion in Plant Biology 2002, 5:94–100
Russell, Peter J. 2012: iGenetics: A Molecular Approach. Pearson Education, Inc., San Francisco, Calif.

Comprehensive Biochemistry for Dentistry pp 495–513 Cite as
Biological Oxidation
- Anil Gupta 2
- First Online: 30 December 2018
1564 Accesses
Please check the hierarchy of section headings, and correct if necessary.
This is a preview of subscription content, log in via an institution .
Buying options
- Available as PDF
- Read on any device
- Instant download
- Own it forever
- Available as EPUB and PDF
- Durable hardcover edition
- Dispatched in 3 to 5 business days
- Free shipping worldwide - see info
Tax calculation will be finalised at checkout
Purchases are for personal use only
Suggested Readings
Alberti KGMN (ed) (1978) Recent advances in clinical biochemistry. Churchill Livingstone, London
Google Scholar
Baron DN (1982) A short textbook of chemical pathology, 4th edn. Wiley, New York
Conn EE, Stump PK (1969) Outline of biochemistry, 2nd edn. Wiley, New Delhi
Harper HA (1979) Review of physiological chemistry, 17th edn. Lange Medical Publisher, New York
Kleiner IS, Orten JM (1966) Biochemistry, 7th edn. Mosby, St Louis
Latner AL (1975) Cantarow and Trumper. Clinical biochemistry, 7th edn. Saunders, Philadelphia
Mazur A, Harrow B (1971) Textbook of biochemistry, 10th edn. Saunders, Philadelphia
McGilvery RW (1983) Biochemistry-a functional approach, 3rd edn. Saunders, Philadelphia
Murray RK, Granner DK, Mayes PA, Rodwell VW (1999) Harper’s biochemistry. Lange Medical Publisher, New York
Murray RK, Granner DK, Mayes PA, Rodwell VW (2003) Harper’s illustrated biochemistry, 26th edn. Lange Medical Books, New York
Oser BL (ed) (1965) Hawk’s: physiological chemistry, 14th edn. Mc-Graw Hill, New York
Rawn JD (1989) Biochemistry. Neil Patterson Publsihers, Burlington, NC
Streyer L (1975) Biochemistry, 3rd edn. Freeman WH, New York
Swaminathan M (1981) Biochemistry for medical students, 1st edn. Geetha Publishers, Mysore
Thorpe WB, Bray HG, James HP (1970) Biochemistry for medical students, 9th edn. Churchill, London
Varley H (1969) Practical clinical biochemistry. WH Medical Books, London
Yudkin M, Offord K (1973) Comprehensive biochemistry. Longman, London
Download references
Author information
Authors and affiliations.
Department of Physiology and Biochemistry, Eklavya Dental College and Hospital, Kotputli, Rajasthan, India
You can also search for this author in PubMed Google Scholar
Rights and permissions
Reprints and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter
Cite this chapter.
Gupta, A. (2019). Biological Oxidation. In: Comprehensive Biochemistry for Dentistry. Springer, Singapore. https://doi.org/10.1007/978-981-13-1035-5_19
Download citation
DOI : https://doi.org/10.1007/978-981-13-1035-5_19
Published : 30 December 2018
Publisher Name : Springer, Singapore
Print ISBN : 978-981-13-1034-8
Online ISBN : 978-981-13-1035-5
eBook Packages : Medicine Medicine (R0)
Share this chapter
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Publish with us
Policies and ethics
- Find a journal
- Track your research
- Subscriber Services
- For Authors
- Publications
- Archaeology
- Art & Architecture
- Bilingual dictionaries
- Classical studies
- Encyclopedias
- English Dictionaries and Thesauri
- Language reference
- Linguistics
- Media studies
- Medicine and health
- Names studies
- Performing arts
- Science and technology
- Social sciences
- Society and culture
- Overview Pages
- Subject Reference
- English Dictionaries
- Bilingual Dictionaries
Recently viewed (0)
- Save Search

Edited by: Richard Cammack , Teresa Atwood , Peter Campbell , Howard Parish , Anthony Smith , Frank Vella , and John Stirling
- Find at OUP.com
- Google Preview
- Share This Facebook LinkedIn Twitter
- View overview page for this topic
Related Content
In this work.
- chemiosmotic coupling hypothesis
Related Overviews
chemiosmotic theory
- Publishing Information
- General Links for this Work
- The Greek alphabet
- Sequence-rule priorities
chemical coupling hypothesis
the hypothesis that the coupling of ATP synthesis to oxidation in oxidative phosphorylation is due to the formation of common ...
Access to the complete content on Oxford Reference requires a subscription or purchase. Public users are able to search the site and view the abstracts and keywords for each book and chapter without a subscription.
Please subscribe or login to access full text content.
If you have purchased a print title that contains an access token, please see the token for information about how to register your code.
For questions on access or troubleshooting, please check our FAQs , and if you can''t find the answer there, please contact us .
- Oxford University Press
PRINTED FROM OXFORD REFERENCE (www.oxfordreference.com). (c) Copyright Oxford University Press, 2023. All Rights Reserved. Under the terms of the licence agreement, an individual user may print out a PDF of a single entry from a reference work in OR for personal use (for details see Privacy Policy and Legal Notice ).
date: 23 April 2024
- Cookie Policy
- Privacy Policy
- Legal Notice
- Accessibility
- [66.249.64.20|91.193.111.216]
- 91.193.111.216
Character limit 500 /500
Resource depletion, pollen coupling, and the ecology of mast seeding
Affiliation.
- 1 Department of Biology, Tufts University, Medford, Massachusetts.
- PMID: 24888210
- DOI: 10.1111/nyas.12465
Masting, the highly variable and synchronous production of seeds across a population of perennial plants, is an ecologically important, but still poorly understood, phenomenon. While much is known about the fitness benefits of masting and its effects on seed consumers and trophic interactions, less is understood about the proximate mechanisms of masting. The resource budget model (RBM) posits that masting requires more resources than plants can gain in a single year. Individual plants store resources until a threshold is reached and then produce seeds, which depletes resources so that plants cannot reproduce again for 2 or more years. Individuals are synchronized by pollen coupling or environmental forcing. We review the assumptions of these models and assess the extent to which they are consistent with general patterns in plant populations. We discuss the implications of the RBM for how plants respond to changes in the external environment. Overall, the RBM is a likely cause of synchrony in many, but not all, masting species. This mechanistic hypothesis also leads to specific, but not always intuitive, expectations about how plant resources affect mast seeding.
Keywords: alternate bearing; mast seeding; nonstructural carbohydrates; pollen coupling; resource budget model.
© 2014 New York Academy of Sciences.
Publication types
- Ecological and Environmental Phenomena*
- Models, Biological
- Pollen / physiology*
- Reproduction
- Seeds / physiology*
COMMENTS
Chemiosmotic coupling hypothesis. Definition. noun. A theory postulated by the biochemist Peter Mitchell in 1961 to describe ATP synthesis by way of a proton electrochemical coupling. Accordingly, hydrogen ion s ( proton s) are pumped from the mitochondrial matrix to the intermembrane space via the hydrogen carrier protein s while the electrons ...
In biological literature, 'coupling' is a common descriptor for a presumed mechanistic link between correlated quantities. The exact nature of such links is often left undefined or unknown, while the term is used in a wide variety of contexts. In single-cell biology, coupling can involve a physical contact between molecular components and/or ligand. At the tissue and organismal level ...
ATP is hydrolyzed to ADP in the following reaction: ATP + H 2 O ⇋ ADP + P i + energy. Note: P i just stands for an inorganic phosphate group (PO 4 3 −) . Like most chemical reactions, the hydrolysis of ATP to ADP is reversible. The reverse reaction, which regenerates ATP from ADP and P i , requires energy.
Furthermore, the definition of the physical species involved in the transfer (proton, hydroxonium ion or proton currents) is still an unresolved issue, even though the latest acquisitions support the idea that protonic currents, difficult to measure, are involved. ... An elegant demonstration of Williams's localized coupling hypothesis came in ...
This is known as a coupling (or cis) configuration. When one wild type allele and one mutant allele are on one homologous chromosome, and the opposite is on the other, this is known as a repulsion (or trans) configuration (e.g., A+b- / a-B+ ). The way to determine the orientation is to look at the parents (or P generation) of that cross if ...
There is good evidence, both direct and indirect, to support the chemiosmotic-coupling hypothesis. Phosphorylation of ADP also occurs in chloroplasts and can be explained by a similar chemiosmotic-coupling mechanism that creates a proton gradient across the organelle's thylakoid membranes. Indeed, A. Jagendorf has shown that a burst of ...
Cell and Molecular Biology Book: Biofundamentals (Klymkowsky & Cooper) 5: Molecular Interactions, Thermodynamics & Reaction Coupling
Genome-wide approaches have extended our understanding of known coupling and coordination mechanisms among eukaryotic gene regulatory processes, and they are revealing previously unknown ...
Coupling in biological systems: Definitions, mechanisms, and implications. Biological systems exhibit an enormous complexity. Their temporal evolution ubiquitously depends on non-linear interactions between non-identical, heterogeneous entities such as molecules, cells, tissues, organs, or organisms. As a result, the observed dynamics at the ...
5: Molecular Interactions, Thermodynamics & Reaction Coupling. While the diversity of organisms and the unique properties of each individual organism are the products of evolutionary processes, initiated billions of years ago, it is equally important to recognize that all biological systems and processes, from growth and cell division to ...
In this section we review theory and data that support the hypothesis of coupling between endogenous genetic barriers and environmental variation. ... but it also explains why species with a similar biology (e.g. similar dispersal abilities) can be affected differently by the same barrier. Often, the natural barrier itself would not reduce gene ...
Quick Reference. A theory concerning oxidative phosphorylation in which it is proposed that the electron-transport chain is arranged such that it generates an energy-rich proton gradient across the inner membrane of a mitochondrion. This energy is then used to drive the phosphorylation of ADP through a membrane-bound ATP-ase. From: chemiosmotic ...
50 years ago Peter Mitchell proposed the chemiosmotic hypothesis for which he was awarded the Nobel Prize for Chemistry in 1978. His comprehensive review on chemiosmotic coupling known as the first "Grey Book", has been reprinted here with permission, to offer an electronic record and easy access to this important contribution to the biochemical literature.
The below mentioned article provides a short note on the Coupling and Repulsion Hypothesis of Gene. Bateson and Punnett in 1906, described a cross in sweat pea, where failure of gene pairs to assort independently was exhibited. Plants of a sweat pea variety having blue flower (BB) and long pollen (LL) were crossed with those of another ...
Figure 7 Coupling phase. In this case, A and B as well as a and b are linked in coupling phase. The non-parental recombinant gametes have the genotype Ab and aB, whereas the parental gametes have the genotype AB and ab. Usually the phenotype cannot be observed in (haploid) gametes, but only in diploid plants.
1 Definition. Biological oxidation is an enzyme-controlled biochemical reaction in which organic molecules of food are oxidized in tissues to release energy. Dietary carbohydrates and lipids are major sources of energy through oxidation. Carbohydrates undergo oxidation in tissues to yield energy along with carbon dioxide and water.
chemical coupling hypothesis Source: Oxford Dictionary of Biochemistry and Molecular Biology Author(s): Richard CammackRichard Cammack, Teresa AtwoodTeresa Atwood, Peter CampbellPeter Campbell, Howard ParishHoward Parish, Anthony SmithAnthony Smith, Frank VellaFrank Vella, John StirlingJohn Stirling
Coupling and Repulsion are both used considering the phenotypic profile of the progeny, and hence, takes into consideration only the distribution of the dominant allele. If, in the progeny of the test cross, the percentage of double dominant is less than that the average percentage of single dominant, that would be what we call repulsion , as ...
We discuss the implications of the RBM for how plants respond to changes in the external environment. Overall, the RBM is a likely cause of synchrony in many, but not all, masting species. This mechanistic hypothesis also leads to specific, but not always intuitive, expectations about how plant resources affect mast seeding.
Furthermore, the definition of the physical species involved in the transfer (proton, hydroxonium ion or proton currents) is still an unresolved issue, even though the latest acquisitions support the idea that protonic currents, difficult to measure, are involved. ... An elegant demonstration of Williams's localized coupling hypothesis came in ...
Individual plants store resources until a threshold is reached and then produce seeds, which depletes resources so that plants cannot reproduce again for 2 or more years. Individuals are synchronized by pollen coupling or environmental forcing. We review the assumptions of these models and assess the extent to which they are consistent with ...