- Sickle cell anemia

On this page
When to see a doctor, risk factors, complications.
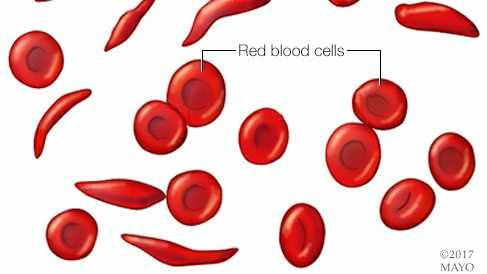
Sickle cell anemia is one of a group of inherited disorders known as sickle cell disease. It affects the shape of red blood cells, which carry oxygen to all parts of the body.
Red blood cells are usually round and flexible, so they move easily through blood vessels. In sickle cell anemia, some red blood cells are shaped like sickles or crescent moons. These sickle cells also become rigid and sticky, which can slow or block blood flow.
The current approach to treatment is to relieve pain and help prevent complications of the disease. However, newer treatments may cure people of the disease.

Red blood cells are usually round and flexible. In sickle cell anemia, some red blood cells look like sickles used to cut wheat. These unusually shaped cells give the disease its name.
Products & Services
- A Book: Mayo Clinic Family Health Book, 5th Edition
- Newsletter: Mayo Clinic Health Letter — Digital Edition
Symptoms of sickle cell anemia usually appear around 6 months of age. They vary from person to person and may change over time. Symptoms can include:
- Anemia. Sickle cells break apart easily and die. Typical red blood cells usually live for about 120 days before they need to be replaced. But sickle cells usually die in 10 to 20 days, leaving a shortage of red blood cells. This is known as anemia. Without enough red blood cells, the body can't get enough oxygen. This causes fatigue.
Episodes of pain. Periodic episodes of extreme pain, called pain crises, are a major symptom of sickle cell anemia. Pain develops when sickle-shaped red blood cells block blood flow through tiny blood vessels to the chest, abdomen and joints.
The pain varies in intensity and can last for a few hours to a few days. Some people have only a few pain crises a year. Others have a dozen or more a year. A severe pain crisis requires a hospital stay.
Some people with sickle cell anemia also have chronic pain from bone and joint damage, ulcers, and other causes.
- Swelling of hands and feet. Sickle-shaped red blood cells block blood circulation in the hands and feet, which can cause them to swell.
- Frequent infections. The spleen is important for protecting against infections. Sickle cells can damage the spleen, raising the risk of developing infections. Babies and children with sickle cell anemia commonly receive vaccinations and antibiotics to prevent potentially life-threatening infections, such as pneumonia.
- Delayed growth or puberty. Red blood cells provide the body with the oxygen and nutrients needed for growth. A shortage of healthy red blood cells can slow growth in babies and children and delay puberty in teenagers.
- Vision problems. Tiny blood vessels that supply blood to the eyes can become plugged with sickle cells. This can damage the portion of the eye that processes visual images, called the retina, and lead to vision problems.
See your healthcare professional right away if you or your child has symptoms of sickle cell anemia, including fever or stroke.
Infections often start with a fever and can be life-threatening. Because children with sickle cell anemia are prone to infections, seek prompt medical attention for a fever greater than 101.5 degrees Fahrenheit (38.5 degrees Celsius).
Seek emergency care for symptoms of stroke, which include:
- One-sided paralysis or weakness in the face, arms or legs.
- Difficulty walking or talking.
- Sudden vision changes.
- Unexplained numbness.
- Severe headache.
From Mayo Clinic to your inbox
Sickle cell anemia is caused by a change in the gene that tells the body to make hemoglobin. Hemoglobin is the iron-rich compound in red blood cells that allows these cells to carry oxygen from the lungs to the rest of the body. The hemoglobin associated with sickle cell anemia causes red blood cells to become rigid, sticky and misshapen.
For a child to have sickle cell anemia, both parents must carry one copy of the sickle cell gene and pass both copies to the child.
If only one parent passes the sickle cell gene to the child, that child will have the sickle cell trait. With one typical hemoglobin gene and one sickle cell gene, people with the sickle cell trait make both typical hemoglobin and sickle cell hemoglobin.
Their blood might contain some sickle cells, but they generally don't have symptoms. They're carriers of the disease. That means they can pass the gene to their children.
For a baby to have sickle cell anemia, both parents must carry a sickle cell gene. In the United States, sickle cell anemia most commonly affects people of African, Mediterranean and Middle Eastern descent.
Sickle cell anemia can lead to a host of complications, including:
- Stroke. Sickle cells can block blood flow to the brain. Signs of stroke include seizures, weakness or numbness of the arms and legs, sudden speech difficulties, and loss of consciousness. If your child has any of these signs or symptoms, seek medical treatment right away. A stroke can be fatal.
- Acute chest syndrome. A lung infection or sickle cells blocking blood vessels in the lungs can cause this life-threatening complication. Symptoms include chest pain, fever and difficulty breathing. Acute chest syndrome might need emergency medical treatment.
- Avascular necrosis. Sickle cells can block the blood vessels that supply blood to the bones. When the bones don't get enough blood, joints may narrow and bones can die. This can happen anywhere but most often happens in the hip.
- Pulmonary hypertension. People with sickle cell anemia can develop high blood pressure in their lungs. This complication usually affects adults. Shortness of breath and fatigue are common symptoms of this condition, which can be fatal.
- Organ damage. Sickle cells that block blood flow to organs deprive the affected organs of blood and oxygen. In sickle cell anemia, blood also is low in oxygen. This lack of oxygen-rich blood can damage nerves and organs, including the kidneys, liver and spleen, and can be fatal.
- Splenic sequestration. Sickle cells can get trapped in the spleen, causing it to enlarge. This may cause abdominal pain on the left side of the body and can be life-threatening. Parents of children with sickle cell anemia can learn how to locate and feel their child's spleen for enlargement.
- Blindness. Sickle cells can block tiny blood vessels that supply blood to the eyes. Over time, this can lead to blindness.
- Leg ulcers. Sickle cell anemia can cause painful open sores on the legs.
- Gallstones. The breakdown of red blood cells produces a substance called bilirubin. A high level of bilirubin in the body can lead to gallstones.
- Priapism. Sickle cell anemia can cause painful, long-lasting erections, known as priapism. Sickle cells can block the blood vessels in the penis, which can lead to impotence over time.
- Deep vein thrombosis. Sickled red blood cells can cause blood clots, increasing the risk of a clot lodging in a deep vein, known as deep vein thrombosis. It also increases the risk of a blood clot lodging in a lung, known as pulmonary embolism. Either can cause serious illness or even death.
- Pregnancy complications. Sickle cell anemia can increase the risk of high blood pressure and blood clots during pregnancy. It also can increase the risk of miscarriage, premature birth and low birth weight babies.
If you carry the sickle cell trait, it can help to see a genetic counselor before you get pregnant. A counselor can help you understand your risk of having a child with sickle cell anemia. You also can learn about possible treatments, preventive measures and reproductive options.
Dec 22, 2023
- Sickle cell disease. National Heart, Lung, and Blood Institute. https://www.nhlbi.nih.gov/health-topics/sickle-cell-disease. Accessed Aug. 4, 2023.
- Field JJ, et al. Overview of the management and prognosis of sickle cell disease. https://www.uptodate.com/contents/search. Accessed Aug. 4, 2023.
- AskMayoExpert. Sickle cell disease. Mayo Clinic; 2022.
- Sickle cell disease (SCD). Centers for Disease Control and Prevention. https://www.cdc.gov/ncbddd/sicklecell/index.html. Accessed Aug. 4, 2023.
- Hoffman R, et al. Pain Management and Antiemetic Therapy in Hematologic Disorders. In: Hematology: Basic Principles and Practice. 8th ed. Elsevier; 2023. https://www.clinicalkey.com. Accessed Aug. 4, 2023.
- Ferri FF. Sickle cell disease. In: Ferri's Clinical Advisor 2024. Elsevier; 2024. https://www.clinicalkey.com. Accessed Aug. 4, 2023.
- Lyfgenia (prescribing information). Bluebird Bio; 2023. https://www.fda.gov/vaccines-blood-biologics/lyfgenia. Accessed Dec. 11, 2023.
- Casgevy (prescribing information). Vertex Pharmaceuticals; 2023. https://www.fda.gov/vaccines-blood-biologics/casgevy. Accessed Dec. 11, 2023.
- Diseases & Conditions
- Sickle cell anemia symptoms & causes
News from Mayo Clinic
More Information
Associated procedures.
- Blood transfusion
- Bone marrow transplant
CON-XXXXXXXX
Make twice the impact
Your gift can go twice as far to advance cancer research and care!
Discussion of Sickle Cell Anemia Essay
Normal hemoglobin-containing erythrocytes are silky, disk-shaped, and supple, resembling doughnuts without holes, and thus readily pass through the blood vessels. Sickle cell hemoglobin-containing cells are stiff and sticky. In oxygen-deficient sickle cells, they take the shape of a sickle or crescent (Alzubaidi et al., 2020). Clot formation is the mechanism through which circulatory components in the plasma become insoluble gel. The gel plugs leak into the blood channels and prevent excessive bleeding. Coagulation factors, calcium, and phospholipids are required for the normal coagulation process. The liver is responsible for the production of coagulation factors.
Sickled cells are not only less elastic but also stickier than healthy red blood cells. The effect can cause blood vessel obstruction, leading to tissue and organ damage with periods of acute pain. The defective blood cells are more brittle and disintegrate, resulting in a lack of erythrocytes, often known as anemia. Sickled red blood cells can obstruct blood supply in veins and capillaries of the brain, resulting in a silent stroke (Hasson et al., 2019). Another prevalent kind of anemia is iron-deficiency anemia, which occurs when the blood lacks sufficient healthy red blood cells. The condition is characterized by reduced or depleted iron reserves necessary for the production of red blood cells.
The most prevalent cause is excessive bleeding. There are no widespread signs and symptoms of the illness. However, people with the condition may suffer from dizziness, weariness, light headaches, a rapid heart rate or palpitations, pale skin, and difficulty breathing. By consuming a diet rich in iron and vitamin C, the condition can be avoided. As such, its treatment encompasses ingestion of vitamins and supplements, which may include iron supplements with a focus on any underlying factors.
Alzubaidi, L., Fadhel, M. A., Al-Shamma, O., Zhang, J., & Duan, Y. (2020). Deep learning models for classification of red blood cells in microscopy images to aid in sickle cell anemia diagnosis . Electronics , 9 (3), 1-18. Web.
Hasson, C., Veling, L., Rico, J., & Mhaskar, R. (2019). The role of hydroxyurea to prevent silent stroke in sickle cell disease: Systematic review and meta-analysis . Medicine, 98 (51), 1-6. Web.
- Chicago (A-D)
- Chicago (N-B)
IvyPanda. (2022, December 7). Discussion of Sickle Cell Anemia. https://ivypanda.com/essays/discussion-of-sickle-cell-anemia/
"Discussion of Sickle Cell Anemia." IvyPanda , 7 Dec. 2022, ivypanda.com/essays/discussion-of-sickle-cell-anemia/.
IvyPanda . (2022) 'Discussion of Sickle Cell Anemia'. 7 December.
IvyPanda . 2022. "Discussion of Sickle Cell Anemia." December 7, 2022. https://ivypanda.com/essays/discussion-of-sickle-cell-anemia/.
1. IvyPanda . "Discussion of Sickle Cell Anemia." December 7, 2022. https://ivypanda.com/essays/discussion-of-sickle-cell-anemia/.
Bibliography
IvyPanda . "Discussion of Sickle Cell Anemia." December 7, 2022. https://ivypanda.com/essays/discussion-of-sickle-cell-anemia/.
- Iron-Deficiency Anemia in Children
- Understanding Sickle Cell Anemia
- Pernicious Anemia: Medical Analysis
- Can the Family Business Disintegrate the Family?
- Blood Vessels - Arteries, Veins, and Capillaries
- Sickle Cell Anemia Medication Effects
- Sickle Cell Anemia: Causes and Treatment
- Disseminated Intravascular Coagulation
- Pathophysiology: The Case Study
- The Sickle Cell Disease Concept
- The Boston Community Analysis in Nursing
- Personal Hygiene: Types and Concept
- Implementation of Telehealth Risk Management Strategy at Fresno Medical Center
- Anaphylactic Shock Symptomology and Treatment
- Pressure Injuries: Assessment and Management for the Interprofessional Team

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- Medicine (Baltimore)
- v.102(47); 2023 Nov 24
- PMC10681612
Psychosocial challenges of persons with sickle cell anemia: A narrative review
Emmanuel aniekan essien.
a Research and Training Unit, Federal Neuropsychiatric Hospital
Blessing F. Winter-Eteng
Chinyere uchechi onukogu, dominic dennis nkangha, faithful miebaka daniel.
b Community and Clinical Research Division, First On-Call Initiative.
Sickle cell anemia (SCA) is a severe form of sickle cell disease that primarily affects black populations and individuals in tropical countries. This condition causes significant morbidity and mortality and leads to a range of psychosocial challenges. A preliminary search was conducted on Ovid Medline and public databases with a combination of Medical Subject Headings keywords, resulting in 368 articles. The articles were screened based on the selection criteria in a nonsystematic method by 3 researchers, and a narrative synthesis was done to analyze extracted data from selected peer-reviewed article. Mental disorders, sleep disturbances, interpersonal relationship challenges, stigmatization, and workplace discrimination were identified as significant contributors to the psychosocial distress experienced by individuals with SCA and their families. Depression and anxiety were prevalent among individuals with SCA, leading to poor treatment adherence, increased pain, and disruptions in various aspects of life. Sleep disturbances, including sleep-disordered breathing and sleepwalking, were also identified as significant contributors to poor sleep quality in SCA patients. Families of individuals with SCA also face challenges, including psychological stress, financial strain, and social disruption. Stigmatization is common, leading to misconceptions and discrimination. Workplace discrimination is prevalent, with a high unemployment rate among adult SCA patients. Comprehensive care is crucial to address these psychosocial issues. Early identification and intervention, comprehensive support programs, patient and family education, enhanced pain management strategies, and integration of mental health into clinical care are recommended. School-based support, research and advocacy, and community support groups are also important. By addressing these challenges through comprehensive care and support, healthcare professionals, policymakers, and society can reduce psychosocial distress and improve the lives of individuals with SCA.
1. Introduction
Sickle cell disease (SCD) is a group of hematological disorders characterized by abnormal hemoglobin, leading to defective red blood cell morphology and function. [ 1 ] The most severe variant of SCD is sickle cell anemia (SCA), caused by an autosomal recessive mutation in the β-globin hemoglobin gene. [ 1 ] The disease has an uneven distribution documented in literature, and much is known about the prevalence of the disease. However, poor early neonatal screening for hemoglobin genotype hampers reliance on data in resource-poor settings. This global disease affects approximately 300,000 babies annually, with 2-thirds of cases occurring in Africa. [ 1 ] Tropical countries like Nigeria, India, and the Democratic Republic of Congo bear the highest burden of SCA. [ 1 ] The pathological consequences of SCA are extensive and affect various aspects of an individual health and have continued to be the subject of research interest. The hallmark of SCA is the abnormal sickling and loss of pliability of red blood cells, leading to vaso-occlusive crises (VOC), hemolytic crises, chronic anemia, and severe pain episodes. [ 2 ] Additionally, individuals with SCA are prone to strokes, recurrent infections, avascular necrosis, priapism, and growth delay. [ 3 ] While advancements in medical interventions have improved life expectancy for SCA patients in high-income countries, the situation remains challenging in low-resource settings. [ 4 ] The lack of healthcare infrastructure and policies in these regions contributes to poorer outcomes for individuals with SCA. [ 4 ]
In addition to the medical syndromes experienced by individuals with SCA, there is an interplay of psychosocial challenges, including mental disorders, pain crises, sleep disorders, interpersonal relationships, substance abuse, stigma, and workplace discrimination, as suggested by literature. [ 5 – 10 ] However, a wider array of psychosocial challenges impacting the patients, their family and peers, and ignored can affect overall response to management, has been underrepresented in literature. [ 11 ] As a result, SCA patients have reported negative experiences due to practitioners ignoring or undermining the effect of these less overt challenges. Such experiences include stigma produced discrediting pain reports, labeling and stereotyping, blaming patients for not improving their health, discrimination, racism, inadequate pain assessment, and delay in care. [ 12 ] On the other hand, there is evidence on the benefits of social support and programs that lead to overall improvement in health-related quality of life (QOL) and coping. [ 13 – 15 ] However, these programs only exist where there is adequate knowledge of the problem and the necessity. Therefore, it is pertinent to review this subject at a global scale to provide an elaborate perspective of the problem and what policies or intervention strategies have proven to be effective and can be domesticated in other climes. This review aims to summarize findings on the prevalence and impact of mental disorders such as anxiety and depression, pain experiences, interpersonal relationships, workplace discrimination, stigma, sleep disorders, and body dissatisfaction among individuals with SCA. These are important factors to consider in addressing psychosocial challenges and improving management strategies. By recognizing and addressing these issues, healthcare providers can prioritize holistic care for SCA patients and improve the overall management and outcomes for individuals living with SCA.
2. Methodology
To address the aims of this review, we attempted answer the following reviewing questions: How prevalent are mental disorders such as anxiety and depression among individuals with SCA, and what is the impact of these disorders on coping mechanisms, interpersonal relationships with peers and family, and overall psychosocial well-being? What is the prevalence and impact of workplace discrimination, stigma, sleep disorders, and body satisfaction among individuals with SCA, and how do these factors influence mental health, substance use, pain experiences, and overall psychosocial challenges?
2.1. Search strategy
A literature search was conducted to identify relevant articles from electronic databases such as PubMed, Embase, PsycINFO, and Scopus. The search strategy included a combination of Medical Subject Headings keywords on Ovid Medline using the following keywords: “anemia, sickle cell,” “mental health,” “social stigma,” “social support,” “quality of life,” “adaptation, psychological,” “mental disorders,” “sickle cell anemia AND mental disorders,” “sleep,” “substance-related disorders,” “pain,” “family relations,” “body dissatisfaction,” “interpersonal relations,” “psychology,” “pain AND psychology,” “medical psychology,” “social stigma OR social support OR quality of life OR sleep OR substance-related disorders OR Family relations OR body dissatisfaction OR interpersonal relations OR (pain AND psychology),” “sickle cell anemia AND social stigma OR social support OR quality of life OR sleep OR substance-related disorders OR Family relations OR body dissatisfaction OR interpersonal relations OR (pain AND psychology),” “social stigma OR social support or psychological adaptation or substance-related disorders or family relations or interpersonal relations or (pain AND psychology),” “sickle cell anemia AND [social stigma OR social support or psychological adaptation or substance-related disorders or family relations or interpersonal relations or (pain AND psychology)].”
2.2. Study selection criteria
Studies were included if they met the following criteria: were peer-reviewed articles, focused on psychosocial issues in SCA, included participants diagnosed with SCA of any age group, published in the English language, and published between 1989 and 2023. Quantitative and qualitative studies were considered for inclusion, including cross-sectional, longitudinal, and intervention studies (Table (Table1 1 ).
Showing article selection process for reviewing psychosocial issues and sickle cell anemia (SCA).
2.3. Data extraction
A data extraction form was developed to collect relevant information from the selected studies. The extracted data included study characteristics (authors, year of publication, study design), participant characteristics (sample size, age, gender), psychosocial issues examined (including depression, anxiety, stigma, QOL, body dissatisfaction, family and peer support), assessment measures used, and key findings related to psychosocial issues in SCA. All Authors were part of the data extraction. The thematic areas were deduced for most themes based on the original authors’ conclusions in the section on other psychosocial issues. All authors read the entire manuscript and decided on the key takeaway.
2.4. Data analysis
A narrative synthesis approach was employed to analyze the extracted data. The findings from the included studies were organized and categorized based on the specific psychosocial issues investigated. Themes and patterns across the studies were identified, and critical results were summarized to provide a comprehensive overview of the psychosocial challenges in SCA patients.
2.5. Ethical considerations
Ethical approval was not required as this study involved the review of existing literature. However, efforts were made to ensure the confidentiality and anonymity of the participants in the original studies by reporting findings in an aggregated and de-identified manner.
2.6. Limitations
Potential limitations of this review include the exclusion of non-English language articles, the reliance on published literature, which may introduce publication bias, and the possibility of missing relevant studies despite the comprehensive search strategy.
Following this methodology, a nonsystematic and comprehensive evaluation of the psychosocial issues in SCA was conducted. A total of 80 research articles were finally selected for the review (Table (Table1). 1 ). The review findings will contribute to a better understanding of the impact of SCA on individuals’ psychosocial well-being, inform healthcare practices, and highlight areas for further research and intervention development (Table (Table2 2 ).
Table showing psychosocial challenges faced by SCA patients and recommendations.
SCA = sickle cell anemia.
4. Discussion
4.1. depression and anxiety in sickle cell anemia.
Mental disorders, such as depression and anxiety, are alarmingly common among SCA individuals, profoundly affecting their overall well-being. These disorders have far-reaching consequences, including increased pain, heightened opioid use, poor treatment adherence, interference with professional duties and schooling, and disruptions in family dynamics. [ 16 , 17 ] Depression, in particular, is most prevalent among patients with SCA, [ 18 ] with studies estimating that globally, 21.6% to 44% of adult SCA patients experience depression. [ 19 ] Similar rates have been observed in the United States, [ 20 ] and Africa, [ 17 ] with a local study in Nigeria finding that nearly 50% of participants experienced depressive states. [ 11 , 21 ] The chronic nature of the disease, the severity of symptoms, and the presence of psychosocial stressors contribute to the high prevalence of depression in this population. [ 22 ] Moreover, depression and anxiety have been shown to predict worse mental and physical health outcomes in SCA patients. [ 17 , 23 – 25 ] A cohort study conducted in Holland revealed that patients with anxiety and depressive disorders experienced severe and disabling chronic pain compared to those without these disorders. [ 17 , 23 – 25 ] These mental disorders significantly impact overall health-related QOL more than the genotype. [ 20 ] Recognizing the detrimental effects of mental disorders on individuals with SCA is crucial for providing comprehensive care. By addressing these mental health challenges, healthcare professionals can enhance the overall management and outcomes for individuals living with SCA.
4.2. Pain among patients with sickle cell anemia
Pain is a prominent feature of SCA that can profoundly impact the lives of individuals with the condition. Unlike typical pain experienced in other conditions, the pain in SCA is characterized by its unpredictable nature and the recurrent episodes of acute pain called VOC. [ 26 ] During VOC, individuals with SCA experience varying degrees of neuropathic pain, including hyperalgesia (increased sensitivity to pain) and allodynia (pain from non-painful stimuli). [ 27 ] Unfortunately, the treatment options for chronic pain in SCA are limited, with opioids being the primary choice. [ 28 ] However, the use of opioids comes with its complications, including constipation, mast cell activation, addiction, and respiratory depression. [ 28 ] Additionally, individuals with SCA often require higher doses of opioids compared to those with other acute or chronic diseases, making pain management more challenging. [ 26 ] The limited efficacy of treating neuropathic chronic pain in SCA may be attributed to the diverse underlying pathophysiology that activates nociceptive fibers. [ 29 ] This includes vascular dysfunction, inflammation, ischemia/reperfusion injury, and oxidative stress. [ 29 ] Pain in SCA can be lifelong, influencing cognitive function and contributing to psychological distress. [ 30 ] Pain in SCA can be lifelong, influencing cognitive function and contributing to psychological distress. [ 28 ] Pain also directly affects emotional expression, behavior, and mood. [ 29 , 30 ] Psychological factors, such as catastrophizing, play a role in pain modulation. [ 31 , 32 ] Catastrophizing refers to an exaggerated negative appraisal of pain, and it can significantly impact pain perception during anticipated or actual pain episodes. [ 33 – 35 ] It involves elements of rumination, magnification, and helplessness. [ 35 ] Catastrophizing behavior is positively correlated with the degree of clinical pain. [ 36 ] In children, higher levels of catastrophizing are associated with an increased risk of disability. [ 37 ] Moreover, higher levels of catastrophizing have been linked to more significant depression and poorer QOL in adults with SCA. [ 34 ] Understanding the complex interplay between pain, psychological factors, and SCA is crucial for effective pain management and improving the overall well-being of individuals with the condition.
4.3. Substance abuse among patients with sickle cell anemia
Chronic pain is a prevalent and debilitating symptom experienced by individuals with SCA. To manage this pain, pain medications, including opioids, are commonly used. However, the addictive properties of these medications can lead to further complications in SCA patients. The severity of pain in SCA can be inferred from the increased use of opioids and the incidence of abuse and addiction among SCA patients. [ 38 ] Analgesics that are commonly abused among individuals with SCA include non-steroidal anti-inflammatory drugs, codeine, oxycodone, and more potent opioids like morphine, levorphanol, methadone, pentazocine, oxymorphone, and fentanyl. [ 38 – 40 ] These medications, while effective in managing pain, can have addictive properties.
A comparative study in Nigeria reported a high incidence of pentazocine addiction among SCA patients who used it for VOC. [ 41 ] In this study, 75% of SCA patients using pentazocine for VOC developed addiction, compared to 15% using other analgesics. [ 41 ] Signs of pentazocine dependence, such as intense drug craving, excessive sweating, non-bone body pains, needle marks, excessive spending, begging, stealing, and poor academic performance, were observed. [ 41 ] These findings highlight the severity of pain experienced by individuals with SCA and the need for alternative pain management strategies to minimize the risks associated with opioid use. It is essential to exercise caution when using pentazocine in SCA patients to prevent addiction and its detrimental consequences. [ 41 ]
Exploring non-opioid pain management options and developing tailored strategies for pain control in SCA are crucial. By identifying alternative approaches and implementing comprehensive pain management plans, healthcare providers can mitigate the risks of opioid addiction while effectively addressing the chronic pain experienced by individuals with SCA. This will ultimately enhance the overall QOL for SCA patients.
4.4. Sickle cell disease and sleep
Sleep is vital to overall well-being, particularly in children and teenagers who require proper rest for their growth and development. [ 42 ] However, various factors can contribute to sleep disturbances, including stress from home and school. [ 43 ] Research has shown that sleep problems are associated with increased physical, mental, and environmental health issues. [ 43 ] In the case of individuals with SCA, episodes of acute pain known as VOC and sleep-disordered breathing have been identified as significant contributors to poor sleep. [ 44 ] Children with SCA have a higher prevalence of sleep-disordered breathing than those without the disease. Studies have reported an increased incidence of obstructive sleep apnea syndrome in children with SCA, surpassing even the prevalence in the general pediatric population. [ 44 ]
Sleep-disordered breathing in SCA can lead to behavioral problems, learning difficulties, elevated blood pressure, bed-wetting, and reduced growth. [ 44 ] These findings are consistent with another study that found a high prevalence of snoring and sleep-disordered breathing in children with SCA aged 2–14. [ 45 ] Interestingly, a survey of Saudi children in the same age group showed even higher prevalence rates of obstructive sleep apnea, snoring, and bed-wetting compared to other countries. [ 46 ] Additionally, there is a significant association between sleep-disordered breathing and sleepwalking in children and adults with SCA. [ 47 ] Adults with SCA also experience a higher prevalence of sleep disorders, and there is an inverse relationship between pain and sleep quality in these patients. [ 22 , 48 ]
Understanding the impact of sleep disturbances in individuals with SCA is crucial for their overall well-being. Sleep-disordered breathing and its associated consequences can profoundly affect cognitive function, behavior, and physical health. Healthcare providers should be aware of these issues and consider incorporating sleep assessments and interventions into the comprehensive care of individuals with SCA. By addressing sleep disturbances, healthcare professionals can potentially improve the QOL and overall health outcomes for individuals living with SCA.
4.5. Interpersonal relationship between individuals with sickle cell anemia and family
Family dynamics play a critical role in supporting adolescents with SCD, providing essential comfort, motivation, and overall support that facilitate effective coping mechanisms and positive relationships with family members and peers. [ 49 , 50 ] However, the challenges associated with SCA can significantly impact families, particularly in tropical countries like Nigeria, where the disease burden is substantial. [ 51 , 52 ]
Economic and psychosocial burdens have been identified as significant stressors in these families, including the inability to meet basic needs, loss of income due to caregiving responsibilities, financial strain related to SCA management, disruption of family interactions, increased conflicts, and neglect of other family members. [ 51 , 52 ]
Partners of adult patients also face unique challenges, such as frequent crises or hospitalizations, psychological stress, financial strain, social disruption, and stigmatization. [ 53 – 56 ] Primary caregivers for children and adolescents with SCA often have limited time for socializing within the family, leading to anxiety and frustration for caregivers and patients. [ 57 ] In this context, mothers primarily serve as caregivers and advocates for their children with SCA. [ 58 ] While familial support is crucial, it can generate problems and tensions. Some parents may attempt to shield their children from social realities, leading to conflicts with adolescents who desire independence and autonomy. [ 58 , 59 ] Adolescent patients may also experience feelings of guilt as they are aware of the physical and economic impact of SCA on their families. [ 59 ] Knowledge about SCA has been found to influence psychological functioning and parent-child dynamics within families positively. [ 49 , 60 ] Families with a comprehensive understanding of the disease tend to have better interpersonal relationships and overall adjustment. Effective coping strategies play a significant role in preventing families from becoming overwhelmed. [ 61 ] Factors such as social support, socioeconomic status, family attitudes, the personality and developmental stage of the child, and other variables can influence coping mechanisms.
Public health education focused on the nature of the disease can enhance coping, improve interpersonal relationships, and positively impact families and society. [ 61 , 62 ] Integrating psycho-educational interventions and psychosocial programs into comprehensive clinical management can provide vital support for SCA patients and their families. [ 63 ] This approach can have reciprocal effects, improving coping in spouses and positively influencing the well-being of SCA patients. [ 63 ]
Healthcare professionals can improve the well-being of individuals with SCA and their families by understanding and addressing family dynamics. Further research and evidence-based interventions are needed to alleviate burdens and promote positive outcomes.
4.6. Interpersonal relationship between individuals with sickle cell anemia and peers
The knowledge and understanding of peers regarding SCA significantly influence the relationships with SCA individuals. [ 64 ] This understanding plays a pivotal role in determining the support and acceptance received by individuals with SCA. [ 64 ] Interestingly, patients with fewer hospitalizations tend to have more positive peer relationships. [ 64 ] Conversely, frequent hospitalizations can lead to a reluctance to form connections, resulting in feelings of isolation and potentially triggering mood disorders. [ 62 ] Regrettably, individuals with SCA often face an elevated risk of bullying and problematic peer relationships, including verbal and physical abuse. [ 50 , 65 , 66 ] This risk is particularly pronounced for males with SCA, who may encounter more aggressive behavior from their non-affected peers. [ 50 ] They may become targets due to their smaller size or be unfairly labeled as lazy when experiencing fatigue. These challenging experiences frequently contribute to social isolation and limited interactions with peers. [ 50 ] As a protective meil puttychanism, individuals with SCA may adapt their behavior to avoid confrontation and potential abuse. [ 50 ]
Promoting understanding among peers is crucial to improving the experiences of individuals with SCA. Increasing awareness and empathy can foster positive relationships and reduce the risk of bullying. Providing support and resources can also contribute to their overall well-being. Further research is needed to develop effective strategies for addressing these issues and promoting positive peer relationships.
4.7. Other psychosocial issues
SCA can significantly impact a person self-image, including how they perceive their body and personality. [ 67 ] This can cause poor body image and emotional problems, which can affect academic performance and achievement in children and adolescents with SCA. [ 65 , 66 , 68 ]
Studies have shown that SCA patients experience higher levels of body dissatisfaction, which correlates with increased stress, interpersonal distrust, and feelings of ineffectiveness. [ 69 , 70 ] Stigmatization is also a common experience for individuals with SCA. In England, 75% of respondents reported stigmatizing incidents, such as being labeled lazy when experiencing fatigue. [ 71 , 72 ] Similar findings were observed in a study conducted in South-West Nigeria, where 70% of subjects reported moderate to high perceived stigma. [ 15 ] Stigmatization can arise from various factors, including using opioids for pain relief, race (being Black), delayed growth/puberty, socioeconomic status, and disease severity. [ 71 ] It can come from unexpected sources such as health institutions, healthcare professionals, family, friends, and society. [ 71 ] The experience of stigma in SCA has been associated with impaired sexuality, higher levels of perceived stress and pain, maladaptive coping, more emergency room visits, poor treatment adherence, and depressive symptoms. [ 73 – 77 ]
Functional impairment is another complication in managing SCA. Neurocognitive deficits have been observed in children and adults with SCA, such as lower intelligence quotient, visuomotor and executive dysfunction, poor working memory, attention and planning difficulties, slower processing speed, language impairments, and deficits in prosodic cues. [ 78 – 81 ] Factors contributing to these deficits include stroke or silent infarcts, anemia severity, malnutrition, cerebral ischemia, and psychosocial factors such as low socioeconomic status, frequent hospitalizations, and family stress. [ 79 , 81 ]
Workplace discrimination is a common experience for SCA patients, with more than half of adult patients being unemployed. [ 82 , 83 ] Siblings of SCA patients without the disease have a significantly higher employability rate than those with the disorder. [ 82 , 84 ] Frequent hospitalizations and educational disruptions can lead to academic under-attainment and job under-qualification. [ 83 , 84 ] For those who secure employment, periodic crises and absenteeism can impede job performance and security, and some SCA patients even report being fired due to discrimination by employers. [ 82 , 83 , 85 ]
Overall, SCA patients generally have a lower QOL, with medical complications, stigmatization, and frequent hospitalizations negatively impacting their health-related QOL. [ 15 ] Studies consistently show poorer QOL in SCA patients than the general population, with significant impairments in physical functioning, emotional roles, social functioning, bodily pain, vitality, and public health perception. [ 15 , 86 , 87 ] To improve the QOL for those with SCA, we need targeted interventions that combat stigma, promote positive self-image, enhance cognitive functioning, and facilitate inclusive and supportive workplaces. Further research is required to develop effective policies and interventions to address the multifaceted impact of SCA on individuals’ lives.
5. Conclusion
SCA is a significant burden, particularly among the black population. Efforts to increase awareness and promote premarital screening are ongoing. However, comprehensive management plans that address the psychosocial challenges associated with the disease are needed. This paper highlights the prevalence of depression, anxiety, substance abuse, sleep disturbances, body dissatisfaction, and peer bullying among individuals with SCA. These challenges, including painful crises, stigma, and economic difficulties, significantly impact treatment response, healthcare costs, and overall QOL. Targeted interventions and holistic care are necessary to address the mental health impact and improve well-being (Table (Table2). 2 ). By recognizing and addressing the psychosocial burden of SCA, we can enhance the QOL and provide comprehensive support. This involves implementing interventions for mental well-being, coping strategies, and addressing unique disease challenges. Holistic care can alleviate psychosocial difficulties and improve the QOL for SCA patients.
6. Recommendation
Comprehensive and holistic care for individuals with SCA includes early identification and intervention, comprehensive support programs, patient and family education, enhanced pain management, integration of mental health into clinical care, school-based support, research and advocacy, community support groups, and continuous improvement (Table (Table2). 2 ). Routine mental health screening should be implemented as part of standard care to identify psychosocial issues early. Comprehensive support programs should be integrated into clinical care, including mental health counseling, peer support groups, and educational workshops on coping strategies, pain management, and disease understanding. A multidimensional approach to pain management should be taken, utilizing non-opioid analgesics, physical therapies, and cognitive-behavioral interventions. Collaboration with schools is necessary to support students with SCA, implementing accommodations and fostering an inclusive school environment. Government investment in research is crucial for understanding the psychosocial burden of SCA and identifying effective interventions while advocating for increased funding and resources. Community engagement efforts should focus on reducing stigma and improving support networks, including establishing community-based support groups. Continuous assessment and evaluation and patient and family feedback will improve support services.
Author contributions
Conceptualization: Emmanuel Aniekan Essien, Faithful Miebaka Daniel.
Data curation: Emmanuel Aniekan Essien, Blessing F. Winter-Eteng, Chinyere Uchechi Onukogu, Dominic Dennis Nkangha, Faithful Miebaka Daniel.
Methodology: Emmanuel Aniekan Essien, Faithful Miebaka Daniel.
Project administration: Emmanuel Aniekan Essien, Faithful Miebaka Daniel.
Resources: Emmanuel Aniekan Essien, Blessing F. Winter-Eteng, Faithful Miebaka Daniel.
Supervision: Emmanuel Aniekan Essien, Faithful Miebaka Daniel.
Validation: Emmanuel Aniekan Essien, Faithful Miebaka Daniel.
Visualization: Faithful Miebaka Daniel.
Writing – original draft: Emmanuel Aniekan Essien, Blessing F. Winter-Eteng, Chinyere Uchechi Onukogu, Dominic Dennis Nkangha, Faithful Miebaka Daniel.
Writing – review & editing: Emmanuel Aniekan Essien, Blessing F. Winter-Eteng, Chinyere Uchechi Onukogu, Dominic Dennis Nkangha, Faithful Miebaka Daniel.
Abbreviations:
The authors have no funding and conflicts of interest to disclose.
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
How to cite this article: Essien EA, Winter-Eteng BF, Onukogu CU, Nkangha DD, Daniel FM. Psychosocial challenges of persons with sickle cell anemia: A narrative review. Medicine 2023;102:47 (e36147).
Home — Essay Samples — Nursing & Health — Sickle Cell Anemia — Sickle Cell Anemia: Types, Symptoms, and Treatment
Sickle Cell Anemia: Types, Symptoms, and Treatment
- Categories: Sickle Cell Anemia
About this sample

Words: 1106 |
Published: Nov 15, 2018
Words: 1106 | Pages: 2 | 6 min read

Cite this Essay
Let us write you an essay from scratch
- 450+ experts on 30 subjects ready to help
- Custom essay delivered in as few as 3 hours
Get high-quality help

Prof Ernest (PhD)
Verified writer
- Expert in: Nursing & Health

+ 120 experts online
By clicking “Check Writers’ Offers”, you agree to our terms of service and privacy policy . We’ll occasionally send you promo and account related email
No need to pay just yet!
Related Essays
6 pages / 2514 words
7 pages / 3056 words
3 pages / 1223 words
3 pages / 1222 words
Remember! This is just a sample.
You can get your custom paper by one of our expert writers.
121 writers online
Still can’t find what you need?
Browse our vast selection of original essay samples, each expertly formatted and styled
Related Essays on Sickle Cell Anemia
Mark Romanek explores the difficult choices that people make when faced with death in his film Never Let Me Go (2010). He explores the raw human emotions of jealousy and forgiveness through the characterisation of Ruth (Keira [...]
Roe vs. Wade was a law established in America on January 22nd, 1973. This court decision allowed women to be able to get abortions in the first trimester. Despite this, the debate on whether abortion should be legal is still [...]
In his Letter to Menoeceus, Epicurus outlines his philosophy of attaining happiness and details the proper attitude that Epicureans should have toward the gods and toward death. In reference to the latter, following his [...]
In 2013, Venka Child aged 16 from Bristol worked with Fixers to create a short video about challenges teen mothers go through. In some part of the video, a teen mother is shown opening a fridge which is almost empty. The teen [...]
Of the dozens of videos you watch every day, how many do you actually remember? The goal of this PSA video is to be one that you would remember. A good PSA is strong, genuine, and powerful enough to leave an impression . To [...]
The Roles of Death and Mortality in “Because I could not stop for Death” Emily Dickinson’s “Because I could not stop for Death” deals with two interrelated yet distinct subjects: death and mortality. The poet presents these [...]
Related Topics
By clicking “Send”, you agree to our Terms of service and Privacy statement . We will occasionally send you account related emails.
Where do you want us to send this sample?
By clicking “Continue”, you agree to our terms of service and privacy policy.
Be careful. This essay is not unique
This essay was donated by a student and is likely to have been used and submitted before
Download this Sample
Free samples may contain mistakes and not unique parts
Sorry, we could not paraphrase this essay. Our professional writers can rewrite it and get you a unique paper.
Please check your inbox.
We can write you a custom essay that will follow your exact instructions and meet the deadlines. Let's fix your grades together!
Get Your Personalized Essay in 3 Hours or Less!
We use cookies to personalyze your web-site experience. By continuing we’ll assume you board with our cookie policy .
- Instructions Followed To The Letter
- Deadlines Met At Every Stage
- Unique And Plagiarism Free

ESSAY SAUCE
FOR STUDENTS : ALL THE INGREDIENTS OF A GOOD ESSAY
Essay: Sickle Cell Anemia
Essay details and download:.
- Subject area(s): Health essays
- Reading time: 3 minutes
- Price: Free download
- Published: 15 November 2017*
- File format: Text
- Words: 818 (approx)
- Number of pages: 4 (approx)
Text preview of this essay:
This page of the essay has 818 words. Download the full version above.
Sickle Cell Anemia is a disease in which someone carries abnormal hemoglobin (Hemoglobin S). Hemoglobin is a protein in red blood cells responsible for carrying oxygen throughout the body, however when people produce Hemoglobin S, it causes the red blood cells to distort into a crescent shape and become fragile, this interrupts healthy blood flow and leads to anemia.
Millions of people worldwide are affected by sickle cell anemia; it is most common among people of Mediterranean, African, Middle Eastern, Asian and Latin American heritage. Each year, about a quarter of a million children are born with the disease. According to the World Health Organisation, approximately 5% of Earth’s population carries trait genes for hemoglobin disorders. The median age of death of male sufferers is 42 years and 48 years for females.
Signs and symptoms of sickle cell anemia vary amongst individuals. One of the most common symptoms are episodes of pain known as “Crisis”, these pains can last for hours to a few weeks. Delayed growth is another symptom of the disease, due to a shortage of healthy red blood cells it causes delayed growth in infants and children and puberty is delayed amongst teenagers. A person may also experience vision problems; this is due to the sickle cells blocking blood vessels in the eye, which can cause harm to the retina. Sickle cells can cause significant harm to an organ that helps to fight off infections; this leaves sufferers prone to frequent infections throughout the body.
About the Genetics:
Sickle cell anemia is due to a mutation in the hemoglobin-Beta gene found on chromosome 11 (one of the 23 pairs of chromosomes in the human body).
Sickle cell anemia is a recessive and inherited condition. In order to have the disease the abnormal hemoglobin S gene must be inherited by both of a person’s parents. If a hemoglobin S gene is inherited from only one parent and a normal hemoglobin gene is inherited from the other parent, their child will have the sickle cell trait but will not be a sufferer of sickle cell anemia.
Detection and Treatment:
Sampling the amniotic fluid surrounding the mother’s womb can test for the sickle cell gene in an unborn baby. In most places, newborn babies can undergo a screening test, which determines the type of hemoglobin each individual produces, any baby who produces hemoglobin S will then be diagnosed with sickle cell anemia. In adults, a blood sample is taken from a vein in the arm, in younger children and infants it is usually drawn from a finger or heel and sent to a laboratory where it is tested for hemoglobin S. If the screening test results are positive, additional tests will be taken to determine how many sickle cell genes are present.
A bone marrow transplant is the only known cure for patients with sickle cell anemia, however it is mainly reserved for people 16 years and younger, as risks increase in older patients. Finding a donor for a bone marrow transplant is extremely difficult and the procedure is of very high-risk, sometimes resulting in death. Even when the operation is successful the body may later on reject the transplant, which leads to harmful complications. Most treatments among sufferers are aimed at reducing and relieving symptoms such as crises and avoiding any possible complications. Blood transfusions are used to increase the number of normal red blood cells, however they carry the risk of iron buildup therefore patients may need further treatment to reduce iron levels. People with sickle cell anemia often take different types of medications such as antibiotics and pain relieving medications such as hydroxyurea.
Life for people living with sickle cell anemia can make it difficult, especially for children who will have to deal with stunted growth and delayed sexual maturity. Patients will have to avoid high altitudes and high-impact exercises in order to prevent a crisis. People describe the feeling of crisis as “repeatedly being stabbed with a butcher knife in the same spot, nonstop.” Most patients will have to deal with fatigue, bacterial infections, progressive tissue and damage to the organs. Patients generally will grow up having to take penicillin daily in order to prevent infections. A sickle cell anemia sufferer’s diet will consist of large portions of protein, vitamins and liquids. New treatments for sickle cell anemia are prolonging life and improving quality of life for sufferers. As recent 45 years ago, the average life expectancy was 14 years old, however now patients can reach over 50.
Bibliography:
Josiah Macy, Oct 5th 2015 http://www.ygyh.org/sickle/whatisit.htm (Date accessed: 5th June 2017)
Mayo Clinic Staff, Dec 29th 2016 http://www.mayoclinic.org/diseases-conditions/sickle-cell-anemia/symptoms-causes/dxc-20303269 (Date accessed: 29th May 2017)
Rev Bras Hematol Hemoter, June 26th 2013 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3832312/ (Date accessed: 7th June 2017)
Sickle Cell Society, July 13th 2014 http://www.sicklecellsociety.org/resources/inheritance-of-sickle-cell-anaemia/ (Date accessed: 7th June 2017)
...(download the rest of the essay above)
About this essay:
If you use part of this page in your own work, you need to provide a citation, as follows:
Essay Sauce, Sickle Cell Anemia . Available from:<https://www.essaysauce.com/health-essays/sickle-cell-anemia/> [Accessed 16-04-24].
These Health essays have been submitted to us by students in order to help you with your studies.
* This essay may have been previously published on Essay.uk.com at an earlier date.
Essay Categories:
- Accounting essays
- Architecture essays
- Business essays
- Computer science essays
- Criminology essays
- Economics essays
- Education essays
- Engineering essays
- English language essays
- Environmental studies essays
- Essay examples
- Finance essays
- Geography essays
- Health essays
- History essays
- Hospitality and tourism essays
- Human rights essays
- Information technology essays
- International relations
- Leadership essays
- Linguistics essays
- Literature essays
- Management essays
- Marketing essays
- Mathematics essays
- Media essays
- Medicine essays
- Military essays
- Miscellaneous essays
- Music Essays
- Nursing essays
- Philosophy essays
- Photography and arts essays
- Politics essays
- Project management essays
- Psychology essays
- Religious studies and theology essays
- Sample essays
- Science essays
- Social work essays
- Sociology essays
- Sports essays
- Types of essay
- Zoology essays
- Undergraduate
- High School
- Architecture
- American History
- Asian History
- Antique Literature
- American Literature
- Asian Literature
- Classic English Literature
- World Literature
- Creative Writing
- Linguistics
- Criminal Justice
- Legal Issues
- Anthropology
- Archaeology
- Political Science
- World Affairs
- African-American Studies
- East European Studies
- Latin-American Studies
- Native-American Studies
- West European Studies
- Family and Consumer Science
- Social Issues
- Women and Gender Studies
- Social Work
- Natural Sciences
- Pharmacology
- Earth science
- Agriculture
- Agricultural Studies
- Computer Science
- IT Management
- Mathematics
- Investments
- Engineering and Technology
- Engineering
- Aeronautics
- Medicine and Health
- Alternative Medicine
- Communications and Media
- Advertising
- Communication Strategies
- Public Relations
- Educational Theories
- Teacher's Career
- Chicago/Turabian
- Company Analysis
- Education Theories
- Shakespeare
- Canadian Studies
- Food Safety
- Relation of Global Warming and Extreme Weather Condition
- Movie Review
- Admission Essay
- Annotated Bibliography
- Application Essay
- Article Critique
- Article Review
- Article Writing
- Book Review
- Business Plan
- Business Proposal
- Capstone Project
- Cover Letter
- Creative Essay
- Dissertation
- Dissertation - Abstract
- Dissertation - Conclusion
- Dissertation - Discussion
- Dissertation - Hypothesis
- Dissertation - Introduction
- Dissertation - Literature
- Dissertation - Methodology
- Dissertation - Results
- GCSE Coursework
- Grant Proposal
- Marketing Plan
- Multiple Choice Quiz
- Personal Statement
- Power Point Presentation
- Power Point Presentation With Speaker Notes
- Questionnaire
- Reaction Paper
- Research Paper
- Research Proposal
- SWOT analysis
- Thesis Paper
- Online Quiz
- Literature Review
- Movie Analysis
- Statistics problem
- Math Problem
- All papers examples
- How It Works
- Money Back Policy
- Terms of Use
- Privacy Policy
- We Are Hiring
Sickle Cell Anemia, Essay Example
Pages: 1
Words: 406
Hire a Writer for Custom Essay
Use 10% Off Discount: "custom10" in 1 Click 👇
You are free to use it as an inspiration or a source for your own work.
Sickle cell anemia is an abnormality found in hemoglobin, a protein found inside red blood cells which delivers oxygen throughout the body. It is genetic in nature and inherited from both mother and father. Red blood cells, which are usually shaped like a disc become sickle or crescent shaped, hence the name. People from tropical, sub-tropical and sub-saharan regions are more likely to have the disease (Wellems et. al. 2496).
Symptoms of sickle cell anemia often start as early as infancy, appearing at about 4 months. Most patients experience painful episodes, referred to as crises and lasting for hours to days at a time (Chiang & Frenette 7782). The pain from these crises is often felt on the bones of the back, chest and long bones. The intensity, duration and frequency of these crises can also vary from patient to patient and can necessitate admission to an acute care facility. Other symptoms associated with sickle cell anemia include fatigue, tachycardia, shortness of breath, paleness and jaundice or yellowing of the eyes and skin. Meanwhile, abdominal pain is largely experienced by younger children. The sickle-shaped cells have less oxygen carrying capacity and they can get trapped more easily within blood vessels, causing blockages and interrupting blood flow. These can result to symptoms such as poor eyesight, blindness, confusion and ulcers, particularly on the lower legs. Growth retardation, delayed sexual development and less than normal body weight is also characteristic findings.
Laboratory tests are crucial in diagnosis and monitoring of the disease. The Hgb S test is often ordered, wherein a blood sample is obtained to check the presence of hemoglobin S. A positive result will necessitate further tests to determine the number of sickle cell genes and differentiate between sickle cell trait and sickle cell disease. Blood sample is further examined under a microscope to verify significant amount of sickle cells to confirm the diagnosis. Other pertinent laboratory tests include a complete blood count, serum creatinine, serum potassium and blood oxygen levels and these are regularly monitored throughout the course of the disease (Clark & Higgins 1284).
Works Cited
Chiang, EY & Frenette, PS. Sickle cell vaso-occlusion. Hematol Oncol Clin North Am . Oct 2005; 19(5):771-84,
Clarke, GM & Higgins, TN. Laboratory investigation of hemoglobinopathies and thalassemias: review and update. Clin. Chem. August 2000 46 (8 Pt 2): 1284–90.
Wellems, TE, Hayton, K, & Fairhurst, RM. The impact of malaria parasitism: from corpuscles to communities. J. Clin. Invest. September 2009; 119 (9): 2496–505.
Stuck with your Essay?
Get in touch with one of our experts for instant help!
Industry Analysis, Term Paper Example
Evaluating Technology, Term Paper Example
Time is precious
don’t waste it!
Plagiarism-free guarantee
Privacy guarantee
Secure checkout
Money back guarantee

Related Essay Samples & Examples
Voting as a civic responsibility, essay example.
Words: 287
Utilitarianism and Its Applications, Essay Example
Words: 356
The Age-Related Changes of the Older Person, Essay Example
Pages: 2
Words: 448
The Problems ESOL Teachers Face, Essay Example
Pages: 8
Words: 2293
Should English Be the Primary Language? Essay Example
Pages: 4
Words: 999
The Term “Social Construction of Reality”, Essay Example
Words: 371
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- My Account Login
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Open access
- Published: 13 April 2024
Multi-center study on mortality in children, and adults with sickle cell anemia-risk factors and causes of death
- Salam Alkindi 1 ,
- Salma Al-Jadidi 1 ,
- Safa Al-Adawi 1 ,
- Refaat Abdullah Elsadek 2 ,
- Ali Al Madhani 3 ,
- Maryam Al-Nabhani 4 &
- Anil V. Pathare 1
Scientific Reports volume 14 , Article number: 8584 ( 2024 ) Cite this article
168 Accesses
Metrics details
- Health care
- Medical research
- Risk factors
Sickle cell disease (SCD) is a major public health burden worldwide with increasing morbidity and mortality. The study evaluates the risk factors associated with mortality in SCD patients, between the years 2006 and 2020 at three hospitals in Oman. The analysis includes clinical manifestations, haematological, biochemical, and radiological parameters, use of antibiotics, and blood and exchange transfusions. Our cohort included 123 patients (82 males, 41 females), with a median age of 27 (Interquartile Range 21–35 years). SCD related complications included acute chest syndrome (ACS) in 52.8%, splenic sequestration in 21.1%, right upper quadrant syndrome in 19.5%, more than > 6 VOC/year in 17.9%, and stroke in 13.8%. At the terminal admission, patients had cough, reduced O 2 saturation, crepitation and fever in 24.4%, 49.6%, 53.6% and 68.3% respectively. Abnormal chest X-ray and chest CT scan were seen in 57.7%, and 76.4% respectively. Laboratory parameters showed a significant drop in hemoglobin (Hb) and platelet counts from baseline, with a significant rise in WBC, LDH and CRP from baseline ( p < 0.05, Wilcoxon Signed Ranks test). All patients received antibiotics, whereas, 95.9% and 93.5% received simple blood transfusions, and exchange transfusions respectively, and 66.6% required non-invasive ventilation. Among the causes of death, ACS is seen in 32 (26%), sepsis in 49 (40%), and miscellaneous in 42 (34%). Sudden death was seen in 32 (26%) of patients. Male gender, with low HbF, rapid drop in Hb and platelet, and increased in WBC, LDH, ferritin, and CRP, correlated significantly with mortality in this cohort.
Similar content being viewed by others

Nationwide retrospective study of critically ill adults with sickle cell disease in France
Maïté Agbakou, Armand Mekontso-Dessap, … Jean-Baptiste Lascarrou

Predictors of impending acute chest syndrome in patients with sickle cell anaemia
Salam Alkindi, Ikhlas Al-Busaidi, … Samir K. Ballas

Risk factors for acute chest syndrome among children with sickle cell anemia hospitalized for vaso-occlusive crises
Faisal A. Alghamdi, Fawaz Al-Kasim, … Rehab Alluqmani
Introduction
Sickle cell disease (SCD) is one of the commonest monogenic diseases with highest prevalence in sun-saharan Africa (500–2000/100.000) 1 . It is caused by a single amino acid mutation, where glutamic acid is replaced by valine in the beta chain of hemoglobin molecule 2 , 3 . The hemoglobin-S polymerizes in post-capillary venules, causing the red blood cells to assume a distorted, sickle-like shape.
Sickle cell disease is a major public health problem in Oman with a marked increase in morbidity and mortality. The estimated sickle cell gene frequency is between 5.8%, and 5.1% with the prevalence of sickle cell trait and SCD is 4.8% and 0.3% respectively 4 , 5 .
Sickle cell disease is characterized by repetitive episodes of vaso-occlusion causing painful episodes, hemolytic anemia, and increased risk of infections impacting severely on survival. Survival estimates however, have continued to improve, with over 95% of SCD patients reaching adulthood, at least in the developed world 6 . In 1994, Platt reported the median survival for patients with HbSS/Sβ 0 thalassemia to be 42–48 years for males and females respectively 7 . This has increased to 53 and 58 years in Jamaica by 2001 8 , and 58 years in the United States in 2014 9 . In recent years, patients are reported to live into their eighth decade 10 . This improvement is attributed to the widespread use of newborn screening programs, penicillin prophylaxis, the universal vaccination program, and the establishment of a comprehensive clinical care program (CCCP) with Hydroxyurea, and blood transfusions 11 .
Although survival rate for children and adults with SCD has improved, however there are still patients who are lost prematurely 11 , 12 , 13 , 14 , 15 . There are many causes of death including sepsis, painful episodes precipitating acute chest syndrome (ACS), and stroke. Previous studies have attempted at identifying risk factors for death including the development of dactylitis, co-inheritance of alpha thalassemia, high Hb F, low baseline Hb, and high WBC count as predictors of severe adverse events 13 , 14 . Some of these were substantiated in subsequent studies, and others were not. The data on specific causes and risk factors for mortality in our country are not available. In a study by Tawfic et al. 16 , ACS was the main cause of ICU admission in patients with SCD. Further, the use of inotropic support and/or mechanical ventilation was an indicator of a high mortality rate among these patients. In another study in children with SCD, Jaiyesimi and Kasem 17 showed that ACS was common irrespective of SCD severity, and all patients appeared to be at risk, but it was increased by Vaso-occlusive crisis (VOC).
Although ACS has remained a leading cause of death, there are no established risk factors for death in patients with SCD in Oman. We are presenting death from three different hospitals in Oman, to identify the main causes of death and its associated risk factors.
Materials and methods
In this retrospective study, 123 SCD patients, who died between 2006 and 2020 at three hospitals in Oman were enrolled, after written approval from the Medical Research Ethics Committee (MREC # 1322), College of Medicine & Health Sciences. All methods were carried out in accordance with relevant guidelines and regulations of the Institution where this research was carried out. Due to the retrospective nature of the study, the need for informed consent was waived by the Medical Research Ethics Committee (MREC #1322). The inclusion criteria included SCD patients of any age, who died between 2006 and 2020. The data was obtained from the electronic patient record system and included demography, the season during death, previous SCD manifestations such as the frequency of painful crisis, ACS, splenic and hepatic sequestration, dactylitis, and stroke. We also analyzed the hematological, biochemical, and radiological parameters at baseline (stable during Outpatient visit), and in the terminal event, as well as the use of antibiotics, ventilatory support, and use of blood /exchange transfusions.
The laboratory results were obtained at baseline and at the terminal event. Hemoglobin and platelet counts were obtained at baseline and nadir, whereas, reticulocyte counts, and white blood cell count (WBC) were recorded at baseline and their maximum. Biochemical parameters included C-reactive protein (CRP), Liver function tests, and Serum LDH and were collected on arrival and at maximum during the terminal event. Additionally, data on basal serum ferritin and HbF were also collected.
Definitions of parameters and SCD complications
To classify the time of death, the year was divided into two seasons based on a mean temperature of 33 °C, between April and August, and a mean temperature of 25 °C between September and March. Stroke was defined as acute neurologic syndrome due to vascular occlusion or hemorrhage in which neurologic symptoms or signs lasted more than 24 h. ACS was defined as the presence of new pulmonary infiltrates on a chest X-ray film, CT scan of the chest, or both, in association with acute respiratory tract symptoms. Sudden death was defined as death from any cause happening within 24 h after hospital admission. Acute splenic sequestration was defined as a decrease from baseline in the hemoglobin level or hematocrit of at least 20 percent, plus a simultaneous increase in the size of the spleen to at least 2 cm below the left costal margin. Right upper quadrant syndrome was defined as SCD painful crisis with signs and symptoms affecting the liver or gallbladder.
Clinical symptoms recorded included temperature, respiratory symptoms, and O 2 saturation by pulse oximetry. Radiology studies including chest X-ray, CT scan and ultra sound of abdomen. Further, SCD therapeutic management protocols like non-invasive ventilatory support, antibiotics, blood transfusion and blood exchange were also recorded. Terminal events were defined as the last hospital admission leading to death of the patient. Causes of death were classified to ACS (clinical & radiological signs), sepsis (clinical, and laboratory evidence with positive microbial culture) and miscellaneous (all others). Miscellaneous causes include cardiac events, pulmonary embolism, stroke, RTA and malignancy.
Statistical analysis
The statistical package for social science (IBM SPSS, USA ver.23, Armonk, NY) was used to analyze the collected data. Normally distributed data were characterized as mean with standard deviation, whereas, data that was not normally distributed was characterized as median with interquartile range (IQR) for continuous variables and percentage and frequency for categorical variables. Wilcoxon Signed Ranks test was used to test the significance of the association between subgroups for various clinical and laboratory parameters. An alpha of < 0.05 was considered to be the statistically significant p value.
123 patients (82 males, 41 females) who were enrolled in the study had a median age (IQR) of 27 (21–35) years, with a range between 1.5 to 79 years. Painful VOC episodes > 6/year were seen in almost one-fifth of this cohort (17.9%), while most patients had a significant past history of SCD-related complications including ACS (52.8%), splenic sequestration (21.1%), right upper quadrant syndrome (19.5%) and stroke (13.8%) (Table 1 ). At the terminal event, fever, cough, abnormal findings in chest examination, and reduced O 2 saturation (pulse oximetry) were seen in 68.3%, 24.4%, 53.6%, and 49.6% respectively.
Amongst the haematology parameters, there was a significant drop in the median hemoglobin and platelet counts from baseline, with a significant rise in the WBC counts ( p < 0.05, Wilcoxon signed ranks test). The median level of baseline hemoglobin (g/dl) dropped significantly from 9.5 to 6.8 g/dl. Reticulocytes were elevated at 5.75%. The median level of white blood cell count (× 10 9 /L) increased significantly to 21 from a baseline level of 11 ( p < 0.05). The median platelet count (× 10 9 /L) dropped significantly to 80 from a baseline level of 319 ( p < 0.05). Amongst the biochemical parameters, the median CRP levels (mg/L) significantly increased from 43 at arrival to the hospital, to the median CRP max value of 171 ( p < 0.05). Similarly, the median serum bilirubin levels (mg/dl) rose significantly from 34 at admission to 105 ( p < 0.05). Further the median serum LDH levels (U/L) also significantly increased from 652 at admission to 1616 at the time of the terminal event ( p < 0.05).
Chest X-ray was abnormal in 71 (57.7%) patients, whereas, the ultrasound showed abnormalities among 54 patients (43.9%). Although a CT scan was done only in 94 patients, it was abnormal in 84 (89.3%) patients. All these patients received antibiotics, whereas, simple and exchange blood transfusions were given in 118 (95.9%) and 115 (93.5%) of patients respectively. Further, 82 patients (66.6%) required ventilatory support.
At the terminal event, 32 (26%) patients died within 24 h of admission to the hospital. 49 (40%) died with sepsis, 32 (26%) died due to ACS associated with multi-organ failure, while the remaining 42 (34%) died due to miscellaneous causes including cardiac events, pulmonary embolism, stroke, RTA, and some died at home. (Table 2 ) Mortality was significantly correlated with a cutoff of basal Hb, basal WBC count, and HbF% of 7 gm/dl, 15 × 10 9 /L, and 8.5% respectively. Leukocytosis was seen in a small number of patients at baseline, but it was significantly high at the time of death. In addition, HbF was a strong predictor of death in all groups. The majority of patients 89 (72.4%) who died had a HbF < 8.5%. About 30% of all the patients, had baseline thrombocytosis, however many of the patients had thrombocytopenia at the time of death. Splenectomy (surgical or auto-splenectomy) was seen in about 50% of all the groups, but no significant differences between all causes of death.
In addition, death due to ACS was significantly seen in the colder months of the year, ( p = 0.04), and sudden death was more likely to happen in ACS, and sepsis group. When we looked at miscellaneous causes of death, cardiac events were most frequent, followed by pulmonary embolism, and stroke. There were a few cases, who died at home, RTA, or cancer (Fig. 1 ).

Details of miscellaneous causes (n, %).
Table 3 shows a comparison of the various risk factors affecting mortality from a literature review. It shows that although survival has improved, patients still die prematurely, especially males; and in this study, the overall proportion of death among males was double that seen in females (66% vs 33%) although the gender ratio in the general SCD Omani patients is 0.51 19 .
Amongst 67 cases with sepsis, 53 (79.1%) were culture-proven, with 11 (20.7%) patients showing multiple microorganisms, while bacterial, fungal, and viral microorganisms were isolated in 32 (60.4%), 6 (11.3%), and 4 (7.5%) respectively (Table 4 ). Among the positive cultures, gram-negative organisms were the commonest, and the highest was seen in the sepsis group, with an increasing number of fungal and viral illnesses. Also, it’s important to note that 16.5% of the cohort had serological evidence of viral hepatitis B, C, and E.
Although survival has improved in SCD, patients still die prematurely, especially males; and in this retrospective study, the overall proportion of death among males was double that seen in females. Similarly, the median age in males was lower (26.5 v/s 28 years), as compared to the females, but this difference was not statistically significant. Comparable findings were also reported in other studies from the region and beyond 14 , 15 , 18 , 20 , 21 .
Karacaoglu et al. 20 , from Turkey, demonstrated that there were more deaths among males, than females, and the mean age of death in their female population was 40.1 years. Although patients are still dying young, data from a French SCD cohort in children under the age of 5 years showed that there is an improvement in survival attributed to the establishment of what is known as complete comprehensive patient care for SCD 21 . Al-Suliman et al. 22 in a study on adults from the eastern province in Saudi Arabia reported a higher prevalence of males 55.8% versus females 44.2% female, with the mean age of the male patients being 30 ± 14 years (range, 16–67 years) and of the female patients 27 ± 13 years (range, 14–67 years). The first Brazilian single institutional study in 2017 showed that the mortality rate was 18.87% among adult SCD patients 23 . Further, combined data between 1997 and 2017, from Brazil showed that mortality was more in the males (50.4%), and patients aged between 25 and 34 years had a higher incidence of deaths 24 . In another study from Brazil between 2000 and 2018, during the entire period the mean ages at death is significantly lower for males, than for females, being 29.4 (± 19.6) versus 33.3 (± 20.3) years respectively 25 . In a study from the UK, the CCU mortality for SCD patients was 19.6% between 2000 and 2007, with the mean ages for males and females respectively being 32.6 and 34.25 years 26 . Further, in a study from the USA with data spanning over 27 years between 1979 and 2005, the mean age at death was significantly different for males (33.4 years) than for females (36.9 years) 27 . Nonetheless, in recent years, patients are reported to live into their eighth decade as reported by Ballas 10 and indeed in this study cohort, we had a SCD patient who died at the age of 79.
Significantly VOC (> 6 episodes/year) were seen in about 20% of cases, reflecting that recurrent VOC remains a marker of severe disease, and these patients accumulate disease-related end-organ damage that leads to their mortality 28 . However, about two-thirds of patients in this cohort (63.4%) had relatively mild disease, indicating the need for intense vigilance in picking up alarm signs for early mortality in some of these relatively mild disease cases.
Many of our patients had a history of ACS, stroke, hepatic & splenic sequestration as well as dactylitis, which were shown to be independent predictors of early childhood mortality in SCD patients 7 , 8 , 9 , 20 . Sixty-five (52.8%) patients had a prior history of ACS, one of the major complications of SCD, and contributed to the terminal event in 25(20.3%) of this cohort. Furthermore, 84 (68.3%) patients presented with fever, cough (24.4%), and 61(49.6%) had reduced O 2 saturation, while 67 (54.5%) had abnormal findings in the chest examination in the terminal episode, with 82 (66.6%) needing ventilatory support. Similarly, 71 (57.7%) patients in this cohort had an abnormal chest X-ray, which was one of the defining features of ACS, and 77.8% had an abnormal CT scan of the chest. Importantly, Knight-Madden et al. 29 , and Tawfiq et al. 16 have shown that mechanical ventilation was found to predict mortality and increased utilization of hospital resources. In this cohort 66.6% needed ventilator assistance, the majority of these patients received simple blood transfusions [118 (95.9%)] and many needed exchange transfusions [115 (93.5%)] as well.
Sepsis seems to be a major contributor to the terminal event as seen in 49 (40%) of cases in this cohort. These patients were characterized by a significantly high CRP, and LDH along with a significant rise in the WBC counts from the baseline, and a significant drop in platelets 30 . Among patients with sepsis, almost 80% of these patients had positive microbial cultures confirming sepsis. These patients received multiple broad-spectrum antibiotics during their terminal event. In the cooperative study of SCD from the USA, only a few bacteria were detected in lung samples at bronchoscopy, possibly since bronchoscopy was performed after empiric antibiotic administration, which is a universal practice 31 . Furthermore, even among SCD patients who had an autopsy done on them, sepsis was identified as the leading cause of death in a postmortem study 32 . More recent paper by Ballas indicated that sepsis contributed to death in a significant number of patients with SCD 33 .
Among the miscellaneous causes, multiorgan failure with sudden death including cardiac events with dilated cardiomayopathy seem to be a leading cause, although pulmonary embolism, stroke and RTA were also other significant causes of mortality in this cohort. Sudden death with bone marrow fat emboli seems to be associated with HbSC and HbSβ + Thalassaemia or the milder SCD phenotypes 34 . However, in this SCD patient cohort HbSβ + Thalassaemia was seen in 22.5% with no HbSC patient and 75.5% patients having HbSS genotype. This is similar to findings from other studies 15 , 18 . Similarly pulmonary embolism (PE), stroke, and RTA were also significant causes of mortality in this cohort. PE is the leading presentation of thromboembolic complications in SCD 35 , 36 , and stroke prevalence was relatively low in our population (probably due to the high prevalence of alpha thalassemia, Alkindi et al. 5 ); however it remains as an important cause of morality. RTA contributed to about 5% of overall total deaths, although no data is available if it was linked to opioid use while driving, on not 33 . Among the miscellaneous also few patients had Bone marrow transplant with GVHD and died with sepsis, and also few patients had malignancy including acute myeloid leukemia, multiple myeloma, sarcoma and myelodysplasia.
Laboratory data showed that there was a statistically significant drop in levels of hemoglobin and platelets count at the terminal event in this study cohort (Table 1 ). A low hemoglobin level has been previously shown to correlate with an increased risk of death with stroke 18 . Although the majority of patients had a baseline Hb > 7 g/dl, the median Hb fell from 9.5 to 6.8, during the terminal event, in all causes of death (Table 2 ). On the contrary, literature reports show that a higher hemoglobin (Hb) level correlated with an increased risk of acute chest syndrome and painful crisis 18 . This was also seen in this study, using the cutoff basal Hb > 7 g/dl and basal WBC > 15 × 10 9 /L (Table 2 ). Consistent with this, many patients in this cohort needed either simple transfusion (95.9%) or exchange transfusions (93.5%).
In a previous prospective study by Plat et al. 7 , the most straightforward laboratory risk factor related to the cause of death was fetal hemoglobin level. Patients with high levels had an improved life expectancy, while adults who had low levels of fetal hemoglobin as children were likely to die earlier than those who had high levels 7 . Thus, when using a cutoff of HbF of 8.5, there was a clear protective effect of high HbF against all types of death, with a significant proportion of these deaths occurring in the low HbF group, ( p < 0.05, Wilcoxon signed ranks test, Table 1 ). This may reflect the heterogeneity of HbS-associated haplotypes in Oman. Although the Arab-Indian haplotype is common in the Eastern provinces in Saudi Arabia 22 in Oman, it only constitutes 25% in the SCD patient population, while Benin and Bantu haplotypes form 50% and 25% respectively 37 . Another, confounding factor related to HbF is hydroxyurea use, which is seen in 36% of this current SCD cohort.
An elevated WBC count was an independent predictor of disease severity as seen in our study. Quinn and Miller 38 in a prospective study reported that leukocytes are known to be involved in the process of vaso-occlusion, and leukocytosis in adults is associated with an increased frequency of ACS and death 39 . Our patients also showed a significant rise in the WBC counts in the terminal event and using a cutoff of 15 × 10 9 /L, it was shown that there was a statistically significant rise in the WBC counts with a median WBC count of 24 and IQR between 20 and 33 (Table 1 ) and this correlated significantly with sepsis during the terminal event.
High serum ferritin was thought to be one of the associated features of mortality among patients with SCD, reflecting both iron overload, as well as inflammatory status. Increased gastrointestinal absorption of iron has been reported in sickle cell disease, because of the associated chronic hemolysis, and anemia. In our study cohort, the median serum ferritin (ng/L) was 1064 with IQR between 323 and 2987. Interestingly, Akinbami et al. 40 , recently reported that 90% of subjects with sickle cell disease had normal iron stores.
Elevated CRP can occur in SCD, during steady state, and in crisis, as reported by us and Okocha et al. 41 , 42 . It represents an underlying inflammatory/ infective, process or tissue necrosis and showed a statistically significant rise, in our cohort, especially among patients who died of sepsis/ multi-organ failure (Table 1 ). In our cohort, the basal median CRP was high (43 mg/L) and showed a statistically significant rise in the terminal episode with a median of 171 ( p < 0.05). Among 67 cases with sepsis, 54 (79.1%) were culture-proven, with 11 (20.7%) patients, showing multiple microorganisms, while bacterial, fungal, and viral microorganisms were isolated in 32 (60.4%), 6 (11.3%), and in 4 (7.5%) respectively. The most frequent organisms were gram-negative organisms, similar to the recent report of the rising gram-negative organisms, reflecting an increasing use of external lines and acquisition of hospital acquired infections 43 .
Our study demonstrated a high serum LDH, high serum bilirubin, and high CRP that further increased significantly toward the terminal event. Several studies have shown these observations and they indicate the degree of inflammation, tissue necrosis, infarction, and haemolysis that is accentuated during the terminal phase 29 , 44 , 45 . Our study demonstrated a statistically significant rise in these parameters in the terminal episode substantiating the above (Table 1 ). These factors with the associated leukocytosis, thrombocytopenia, and drop in hemoglobin heralded the onset of multiorgan failure in this cohort.
The cause of death was established by the course of illness during hospitalization, supportive laboratory evidence and telephonic enquiries with relatives in case of out of hospital deaths of these patients. Although ACS has remained a leading cause of death, there are no established risk factors for death in patients with SCD in Oman. In a study from Oman by Tawfic et al. 16 ACS was the main cause of ICU admission in patients with SCD. Further, the use of inotropic support and/or mechanical ventilation was an indicator of high mortality rate among these patients. Jaiyesimi and Kasem 17 showed that ACS was common irrespective of SCD severity in children from Oman, and all patients appeared to be at risk, but it was increased in patients with vaso-occlusive crisis.
When we looked at the time of death, we observed increased mortality due to ACS and sepsis during the colder months of the year (September to March). This is not unusual as viral infections such as respiratory syncytial virus and seasonal influenza predominate during that period and it is known to be one of the leading viral causes of ACS. Our own data and others showed similar patterns 46 , 47 . We also observed that over 30% of deaths occurred within 24 h of hospital admission. This data is also similar to the previous studies and the highest numbers were reported in the ACS and sepsis groups 48 . In the absence of post-mortem studies, (which are rarely accepted by the patient’s relatives), the exact cause will remain elusive or putative, although the recent literature is suggestive of increased cardiovascular events in this situation.
SCD is a complex condition, with life-threatening complications that interfere with the patient’s normal life owing to repeated painful episodes, and the associated multi-system involvement. Although this is a retrospective study, however, it identified that ACS and sepsis were the most important preventable risk factor associated with mortality in this SCD patient cohort. A high index of suspicion and vigilance is required for patients presenting with fever, tachycardia, and signs of ACS, associated with a drop in Hb, with raised WBC, CRP, LDH, and ferritin, to identify these patients early and initiate appropriate rapid management protocols.
In summary, the ability to identify the risk factors that are associated with increased mortality among SCD patients permits accurate prognostication and provides effective prophylactic management strategies. This study shows that male gender, low HbF, substantial drop in hemoglobin and platelet, as well as increased in WBC counts, serum LDH, ferritin and CRP, correlated significantly with mortality risk during the terminal event in patients with SCD.
Data availability
Data is available on request from the authors. The corresponding author can be contacted for details of the study.
Colombatti, R., Birkegard, C. & Medici, M. PB2215—Global epidemiology of sickle cell disease: A systematic review. HemaSphere 6 , 2085–2086. https://doi.org/10.1097/01.HS9.0000851688.00394.f4 (2022).
Article PubMed Central Google Scholar
Ware, R. E., de Montalembert, M., Tshilolo, L. & Abboud, M. R. Sickle cell disease. Lancet 390 (10091), 311–323. https://doi.org/10.1016/S01406736(17)30193-9 (2017).
Article PubMed Google Scholar
Bunn, H. F. Pathogenesis and treatment of sickle cell disease. N. Engl. J. Med. 337 (11), 762–769. https://doi.org/10.1056/NEJM199709113371107 (1997).
Article CAS PubMed Google Scholar
Al-Riyami, A. A., Suleiman, A. J., Afifi, M., Al-Lamki, Z. M. & Daar, S. A community-based study of common hereditary blood disorders in Oman. East Mediterr. Health J. 7 (6), 1004–1011 (2001).
Alkindi, S. et al. Forecasting hemoglobinopathy burden through neonatal screening in Omani neonates. Hemoglobin. 34 (2), 135–144. https://doi.org/10.3109/03630261003677213 (2010).
Telfer, P. et al. Clinical outcomes in children with sickle cell disease living in England: A neonatal cohort in East London. Haematologica 92 (7), 905–912. https://doi.org/10.3324/haematol.10937 (2007).
Platt, O. S. et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N. Engl. J. Med. 330 (23), 1639–1644. https://doi.org/10.1056/NEJM199406093302303 (1994).
Wierenga, K. J., Hambleton, I. R. & Lewis, N. A. Survival estimates for patients with homozygous sickle-cell disease in Jamaica: A clinic-based population study. Lancet. 357 (9257), 680–683. https://doi.org/10.1016/s0140-6736(00)04132-5 (2001).
Elmariah, H. et al. Factors associated with survival in a contemporary adult sickle cell disease cohort. Am. J. Hematol. 89 (5), 530–535. https://doi.org/10.1002/ajh.23683 (2014).
Article CAS PubMed PubMed Central Google Scholar
Ballas, S. K., Pulte, E. D., Lobo, C. & Riddick-Burden, G. Case series of octogenarians with sickle cell disease. Blood. 128 (19), 2367–2369. https://doi.org/10.1182/blood-2016-05-715946 (2016).
Quinn, C. T., Rogers, Z. R., McCavit, T. L. & Buchanan, G. R. Improved survival of children and adolescents with sickle cell disease. Blood 115 (17), 3447–3452. https://doi.org/10.1182/blood-2009-07-233700 (2010).
Vichinsky, E., Hurst, D., Earles, A., Kleman, K. & Lubin, B. Newborn screening for sickle cell disease: Effect on mortality. Pediatrics. 81 (6), 749–755 (1988).
Yanni, E., Grosse, S. D., Yang, Q. & Olney, R. S. Trends in pediatric sickle cell disease-related mortality in the United States, 1983–2002. J. Pediatr. 154 (4), 541–545. https://doi.org/10.1016/j.jpeds.2008.09.052 (2009).
Vichinsky, E. P. et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group. N. Eng. J. Med. 342 (25), 1855–1865. https://doi.org/10.1056/NEJM200006223422502 (2000).
Article CAS Google Scholar
Leikin, S. L. et al. Mortality in children and adolescents with sickle cell disease. Cooperative Study of Sickle Cell Disease. Pediatrics 84 (3), 500–508 (1989).
Tawfic, Q. A. et al. Adult sickle cell disease: A five-year experience of intensive care management in a university hospital in Oman. Sultan Qaboos Univ. Med. J. 12 (2), 177–183. https://doi.org/10.12816/0003110 (2012).
Article PubMed PubMed Central Google Scholar
Jaiyesimi, O. & Kasem, M. Acute chest syndrome in Omani children with sickle cell disease: Epidemiology and clinical profile. Ann. Trop. Paediatr. 27 (3), 193–199. https://doi.org/10.1179/146532807x220307 (2007).
Miller, S. T. et al. Prediction of adverse outcomes in children with sickle cell disease. N. Engl. J. Med. 342 (2), 83–89. https://doi.org/10.1056/NEJM200001133420203 (2000).
https://data.gov.om/OMPOP2016/population?region=1000010&indicator=1000250&nationality=1000000&sex=1000000&age-group=1000000&frequency=A .
Karacaoglu, P. K. et al. East Mediterranean region sickle cell disease mortality trial: Retrospective multicenter cohort analysis of 735 patients. Ann. Hematol. 95 (6), 993–1000. https://doi.org/10.1007/s00277-016-2655-5 (2016).
Desselas, E. et al. Mortality in children with sickle cell disease in mainland France from 2000 to 2015. Haematologica 105 (9), e440-443. https://doi.org/10.3324/haematol.2019.237602 (2020).
Al-Suliman, A. et al. Patterns of mortality in adult sickle cell disease in the Al-Hasa region of Saudi Arabia. Ann. Saudi Med. 26 (6), 487–488. https://doi.org/10.5144/0256-4947.2006.487 (2006).
de Castro Lobo, C. L. et al. Mortality in children, adolescents and adults with sickle cell anemia in Rio de Janerio, Brazil. Hematol. Transfus. Cell Ther. 40 (1), 37–42. https://doi.org/10.1016/j.bjhh.2017.09.006 (2018).
Article Google Scholar
Mota, F. M. et al. Analysis of the temporal trend of mortality from sickle cell anemia in Brazil. Rev. Bras. Enferm. 75 (4), e20210640. https://doi.org/10.1590/0034-7167-2021-0640 (2022).
Santo, A. H. Sickle cell disease related mortality in Brazil, 2000–2018. Hematol. Transfus. Cell Ther. 44 (2), 177–185. https://doi.org/10.1016/j.htct.2020.09.154 (2022).
Gardner, K. et al. Outcome of adults with sickle cell disease admitted to critical care—Experience of a single institution in the UK. Br. J. Hematol. 150 , 610–613. https://doi.org/10.1111/j.1365-2141.2010.08271.x (2010).
Lanzkron, S., Carroll, C. P. & Haywood, C. Mortality rates and age at death from sickle cell disease: US, 1979–2005. Public Health Rep. 128 , 110–116. https://doi.org/10.1177/003335491312800206 (2013).
Darbari, D. S. et al. Severe painful vaso-occlusive crises and mortality in a contemporary adult sickle cell anemia cohort study. PLoS One. 8 (11), e79923. https://doi.org/10.1371/journal.pone.0079923 (2013).
Article ADS CAS PubMed PubMed Central Google Scholar
Knight-Madden, J. M., Barton-Gooden, A., Weaver, S. R., Reid, M. & Greenough, A. Mortality, asthma, smoking and acute chest syndrome in young adults with sickle cell disease. Lung. 191 (1), 95–100. https://doi.org/10.1007/s00408-012-9435-3 (2013).
Shome, D. K. et al. The platelet count and its implications in sickle cell disease patients admitted for intensive care. Indian J. Crit. Care Med. 22 (8), 585–590. https://doi.org/10.4103/ijccm.IJCCM_49_18 (2018).
Castro, O. et al. The acute chest syndrome in sickle cell disease: Incidence and risk factors. The Cooperative Study of Sickle Cell Disease. Blood. 84 (2), 643–649 (1994).
Manci, E. A. et al. Causes of death in sickle cell disease: An autopsy study. Br. J. Haematol. 123 (2), 359–365. https://doi.org/10.1046/j.1365-2141.2003.04594.x (2003).
Ballas, S. K. Opioids are not a major cause of death of patients with sickle cell disease. Ann. Hematol. 100 (5), 1133–1138. https://doi.org/10.1007/s00277-021-04502-2 (2021).
Tsitsikas, D. A. et al. Revisiting fat embolism in sickle syndromes: Diagnostic and emergency therapeutic measures. Br. J. Haematol. 186 , e112–e115. https://doi.org/10.1111/bjh.15941 (2019).
Naik, R. P., Streiff, M. B., Haywood, C., Segal, J. B. & Lanzkron, S. Venous thromboembolism incidence in the cooperative study of sickle cell disease. J. Thromb. Haemost. 12 (12), 2010–2016. https://doi.org/10.1111/jth.12744 (2014).
Alkindi, S. et al. Predicting risk factors for thromboembolic complications in patients with sickle cell anaemia—Lessons learned for prophylaxis. J. Int. Med. Res. 49 (12), 3000605211055385. https://doi.org/10.1177/03000605211055385 (2021).
Daar, S., Hussain, H. M., Gravell, D., Nagel, R. L. & Krishnamoorthy, R. Genetic epidemiology of HbS in Oman: Multicentric origin for the βS gene. Am J Hematol 64 (1), 39–46. https://doi.org/10.1002/(sici)1096-8652(200005)64:1%3c39::aid-ajh7%3e3.0.co;2-# (2000).
Quinn, C. T. & Miller, S. T. Risk factors and prediction of outcomes in children and adolescents who have sickle cell anemia. Hematol. Oncol. Clin. N. Am. 18 (6), 1339–1354. https://doi.org/10.1016/j.hoc.2004.07.004 (2004).
Article MathSciNet Google Scholar
Alkindi, S. et al. Predictors of impending acute chest syndrome in patients with sickle cell anaemia. Sci. Rep. 10 (1), 2470. https://doi.org/10.1038/s41598-020-59258-y (2020).
Akinbami, A. A. et al. Serum ferritin levels in adults with sickle cell disease in Lagos, Nigeria. J. Blood Med. 4 , 59–63. https://doi.org/10.2147/JBM.S42212 (2013).
Pathare, A. et al. Cytokine profile of sickle cell disease in Oman. Am. J. Hematol. 77 (4), 323–328. https://doi.org/10.1002/ajh.20196 (2004).
Okocha, C. et al. C-reactive protein and disease outcome in Nigerian sickle cell disease patients. Ann. Med. Health Sci. Res. 4 (5), 701–705. https://doi.org/10.4103/2141-9248.141523 (2014).
Al-Tawfiq, J. A., Rabaan, A. A. & AlEdreesi, M. H. Frequency of bacteremia in patients with sickle cell disease: A longitudinal study. Ann. Hematol. 100 (6), 1411–1416. https://doi.org/10.1007/s00277-021-04523-x (2021).
Maitre, B. et al. Acute chest syndrome in adults with sickle cell disease. Chest. 117 (5), 1386–1392. https://doi.org/10.1378/chest.117.5.1386 (2000).
Allareddy, V. et al. Outcomes of acute chest syndrome in adult patients with sickle cell disease: Predictors of mortality. PLoS One 9 (4), e94387. https://doi.org/10.1371/journal.pone.0094387 (2014).
Sadreameli, S. C., Reller, M. E., Bundy, D. G., Casella, J. F. & Strouse, J. J. Respiratory syncytial virus and seasonal influenza cause similar illnesses in children with sickle cell disease. Pediatr. Blood Cancer. 61 (5), 875–878. https://doi.org/10.1002/pbc.24887 (2014).
Alkindi, S., Al-Yahyai, T., Raniga, S., Boulassel, M. R. & Pathare, A. Respiratory viral infections in sickle cell anemia: Special emphasis on H1N1 co-infection. Oman Med. J. 35 (6), e197. https://doi.org/10.5001/omj.2020.89 (2020).
Nze, C. et al. Sudden death in sickle cell disease: Current experience. Br. J. Haematol. 188 (4), e43–e45. https://doi.org/10.1111/bjh.16314 (2020).
Download references
Acknowledgements
We wish to thank Professor Samir K. Ballas for his constructive comments on the manuscript, and the hospital administration for allowing the use of hospital material in this study.
Author information
Authors and affiliations.
Department of Haematology, College of Medicine and Health Sciences, Sultan Qaboos University, P. O. Box 35, 123, Muscat, Oman
Salam Alkindi, Salma Al-Jadidi, Safa Al-Adawi & Anil V. Pathare
Department of Medicine, Nizwa Hospital, Nizwa, Oman
Refaat Abdullah Elsadek
Department of Medicine, Sohar Hospital, Sohar, Oman
Ali Al Madhani
Emergency Department, Royal Hospital, Muscat, Oman
Maryam Al-Nabhani
You can also search for this author in PubMed Google Scholar
Contributions
All authors have made substantial contributions and have seen and approved the final version of manuscript. (1) S.A.K., S.A.J., S.A.A., R.A.E. and A.A.M. were fully involved in the conception and design of the study, acquisition, analysis and interpretation of data, (2) S.A.K., and A.V.P. were instrumental in the drafting the article and critical appraisal before submission.
Corresponding author
Correspondence to Salam Alkindi .
Ethics declarations
Competing interests.
The authors declare no competing interests.
Additional information
Publisher's note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ .
Reprints and permissions
About this article
Cite this article.
Alkindi, S., Al-Jadidi, S., Al-Adawi, S. et al. Multi-center study on mortality in children, and adults with sickle cell anemia-risk factors and causes of death. Sci Rep 14 , 8584 (2024). https://doi.org/10.1038/s41598-024-58328-9
Download citation
Received : 20 June 2023
Accepted : 27 March 2024
Published : 13 April 2024
DOI : https://doi.org/10.1038/s41598-024-58328-9
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Sickle cell disease
- Predictors of mortality
- Acute chest syndrome
- Sudden death
By submitting a comment you agree to abide by our Terms and Community Guidelines . If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing newsletter — what matters in science, free to your inbox daily.

Well-planned online essay writing assistance by PenMyPaper
Writing my essays has long been a part and parcel of our lives but as we grow older, we enter the stage of drawing critical analysis of the subjects in the writings. This requires a lot of hard work, which includes extensive research to be done before you start drafting. But most of the students, nowadays, are already overburdened with academics and some of them also work part-time jobs. In such a scenario, it becomes impossible to write all the drafts on your own. The writing service by the experts of PenMyPaper can be your rescuer amidst such a situation. We will write my essay for me with ease. You need not face the trouble to write alone, rather leave it to the experts and they will do all that is required to write your essays. You will just have to sit back and relax. We are offering you unmatched service for drafting various kinds for my essays, everything on an online basis to write with. You will not even have to visit anywhere to order. Just a click and you can get the best writing service from us.
Testimonials
My experience here started with an essay on English lit. As of today, it is quite difficult for me to imagine my life without these awesome writers. Thanks. Always.
Professional essay writing services
Customer Reviews
Connect with the writers
Once paid, the initial draft will be made. For any query r to ask for revision, you can get in touch with the online chat support available 24X7 for you.
IMAGES
VIDEO
COMMENTS
What is sickle cell? Sickle cell disease, also called sickle cell anemia or just sickle cell, is a genetic disease where red blood cells can take the shape of a crescent or a sickle, and that change allows them to be more easily destroyed- causing anemia, among other things. Sickle cell disease is caused by defective hemoglobin, which is the ...
Introduction. The essay is an in depth critical examination-a quantitative critique of two article based on sickle cell and anemia titled "Development and Evaluation of a Sickle Cell Disease Assessment Instrument" and "Iron Deficiency Anemia: Following Prenatal Nutrition Interventions" written by Day Sara and Leblanc Caroline et al ...
Researching the Sickle Cell Anaemia Essay. Sickle cell anaemia is a hereditary disease passed down from parents to their children. The red blood cells, which are typically disc-shaped, take an abnormal shape and form a crescent or sickle form. The disease shortens the lifespan of the people infected by weakening the body's immunity system ...
Exploring Sickle Cell Anemia through essay writing provides an opportunity to deepen understanding, raise awareness, and contribute to the advancement of research and support for individuals and families affected by this condition. By choosing meaningful and engaging topics, writers can make a valuable contribution to the discourse surrounding ...
Sickle cell anemia is caused by a change in the gene that tells the body to make hemoglobin. Hemoglobin is the iron-rich compound in red blood cells that allows these cells to carry oxygen from the lungs to the rest of the body. The hemoglobin associated with sickle cell anemia causes red blood cells to become rigid, sticky and misshapen.
Sickle cell disease (SCD), or sickle cell anemia, is a group of genetic conditions, resulting from the inheritance of a mutated form of the gene coding for the β globulin chain of the hemoglobin molecule, which causes malformation of red blood cells (RBCs) in their deoxygenated state. Specifically, this single point mutation occurs at position ...
Sickle cell anemia is an inherited disorder of the globin chains that causes hemolysis and chronic organ damage. Sickle cell anemia is the most common form of sickle cell disease (SCD), with a lifelong affliction of hemolytic anemia requiring blood transfusions, pain crises, and organ damage. Since the first description of the irregular sickle ...
Sickle cell anemia is a disease that deals with hemoglobin, the protein that aids in oxygen transport from organ to organ, in red blood cells. Normally red blood cells have a biconcave shape that attributes to a maximum capacity of hemoglobin, but in cases of sickle cell anemia, red blood cells are structured, fragile, and crescent-shaped.
Anyone who has sickle cell anemia is at risk for stroke, including babies. Approximately 11% of people with sickle cell anemia have strokes by age 20, and 24% have strokes by age 45. Here is information on stroke symptoms: Severe headache. Sudden weakness on one side of your or your child's body. Change in alertness.
INTRODUCTION. In 1910, sickle cell disease burst onto the Western medical scene as a "strange" or, as Herrick termed it, a "new, unknown disease." 1 Physicians were intrigued by the sickled appearance of the red cells in this disorder, and case reports and analytical papers detailing the clinical features of this disorder appeared to almost always involve people of color. 2-6 The ...
Sickle cell anemia was the first genetic disease that caused by point mutation in haemoglobin gene and produce the abnormal haemoglobin S (Hb S). This genetic disease is very common among African descent, about 8% of African-American descents have sickle cell anemia in United State, while in Africa, approximately 1 in 100 people will have this ...
Discussion of Sickle Cell Anemia Essay. Normal hemoglobin-containing erythrocytes are silky, disk-shaped, and supple, resembling doughnuts without holes, and thus readily pass through the blood vessels. Sickle cell hemoglobin-containing cells are stiff and sticky. In oxygen-deficient sickle cells, they take the shape of a sickle or crescent ...
Essay on Sickle Cell Anemia. Sickle Cell Anemia is a hereditary disease that changes the smallest and most important components of the body. A gene causes the bone marrow in the body to make sickled shapes, when this happens; it causes the red blood cell to die faster. This is what causes Hemolytic Anemia. Older children and adults with sickle ...
1. Introduction. Sickle cell disease (SCD) is a group of hematological disorders characterized by abnormal hemoglobin, leading to defective red blood cell morphology and function. [] The most severe variant of SCD is sickle cell anemia (SCA), caused by an autosomal recessive mutation in the β-globin hemoglobin gene. [] The disease has an uneven distribution documented in literature, and much ...
Sickle Cell Anemia Essay. Sickle Cell Anemia Sickle cell anemia is caused by a defect in the gene that controls the production of normal hemoglobin, which is an iron-containing protein in red blood cells that transports oxygen from the lungs to body tissues. The defective gene results in the production of abnormal hemoglobin known as ...
Sickle Cell Anemia essay example for your inspiration. ️ 1838 words. Read and download unique samples from our free paper database. ... Sickle cell anemia is a hereditary condition in which the red blood cells have the crescent or sickle shape instead of the ordinary oval shape. The Pathophysiology, signs and symptoms, diagnosis, and ...
Sickle cell anemia represents one of the various blood disorders categorized as sickle cell disease (SCD). It is a life-threatening condition in the absence of comprehensive treatment due to its diverse impact on the functional capability of the cardiovascular/blood organ systems. Inusa et al. (2019)
Sickle cell anemia, or sickle cell disease, is a genetic disease and red blood cells that are normally shaped like a disc have instead a crescent shape. This causes them to have a difficult time going through small vessels throughout the body. A mutation on the hemoglobin-Beta gene on chromosome 11 causes sickle cell anemia.
Sickle cell anemia is due to a mutation in the hemoglobin-Beta gene found on chromosome 11 (one of the 23 pairs of chromosomes in the human body). Sickle cell anemia is a recessive and inherited condition. In order to have the disease the abnormal hemoglobin S gene must be inherited by both of a person's parents.
Sickle cell anemia is an abnormality found in hemoglobin, a protein found inside red blood cells which delivers oxygen throughout the body. It is genetic in nature and inherited from both mother and father. Red blood cells, which are usually shaped like a disc become sickle or crescent shaped, hence the name. People from tropical, sub-tropical ...
Sickle Cell Anemia Essay. 401 Words2 Pages. Sickle cell anemia is one of the many kinds of anemia that you can have. This specific type of anemia affects more than 72,000 people in the U.S alone. Sickle cell anemia is a type of red blood disorder that causes the body to make hemoglobin S it can also cause a person to feel less energized.
Sickle cell disease (SCD) is one of the commonest monogenic diseases with highest prevalence in sun-saharan Africa (500-2000/100.000) 1. It is caused by a single amino acid mutation, where ...
1. Sickel Cell Anemia Essay examples Sickle Cell anemia is a group of inherited red blood cell disorders, or a collection of recessive genetic disorders characterized by a hemoglobin variant called Hb S. Normal red blood cells are round like doughnuts, and they move through small blood tubes in the body to deliver oxygen. Sickle red blood cells become hard, sticky and shaped like sickles used ...
Also, in search for an above-average essay writing quality, more means better, whereas content brought by a native English speaker is always a smarter choice. So, if your budget affords, go for one of the top 30 writers on our platform. ... Essay Sickle Cell Anemia, Personal Essay Writer For Hire, Essays On Health, Best Creative Essay Writer ...